第17章 Graphics模块中的基因组可视化包—GenomeDiagram¶
Bio.Graphics 模块基于Python的第三方扩展库 ReportLab ,ReportLab主要生成PDF文件,同时也能生成EPS(Encapsulated Postscript)文件和SVG文件。ReportLa可以导出矢量图,如果安装依赖关系(Dependencies),比如 PIL(Python Imaging Library) ,ReportLab也可以导出JPEG, PNG, GIF, BMP和PICT格式的位图(Bitmap image)。
17.1 基因组可视化包—GenomeDiagram¶
17.1.1 GenomeDiagram简介¶
Bio.Graphics.GenomeDiagram 包被整合到Biopython 1.50版之前,就已经是Biopython的独立模块。GenomeDiagram包首次出现在2006年Pritchard等人在Bioinformatics杂志的一篇文章 [2] ,文中展示了一些图像示例,更多图像示例请查看GenomeDiagram手册 http://biopython.org/DIST/docs/GenomeDiagram/userguide.pdf 。正如“GenomeDiagram”名称所指,它主要用于可视化全基因组(特别是原核生物基因组),即可绘制线型图也可绘制环形图,Toth等人在2006年发表的文章 [3] 中图2就是一个示例。Van der Auwera 等人在2009年发表的文章 [4] 中图1和图2也进一步说明,GenomeDiagram适用于噬菌体、质粒和线粒体等微小基因组的可视化。
如果存储基因组信息的是从GenBank文件中下载的 SeqRecord 话,它会包含许多 SeqFeature ,那么用这个模块处理就很简单(详见
第 4 章和第 5 章)。
17.1.2 图形,轨迹, 特征集和特征¶
GenomeDiagram使用一组嵌套的对象,图层中沿着水平轴或圆圈的图形对象(diagram object)表示一个序列(sequence)或序列区域(sequence region)。一个图形可以包含多个轨迹(track),呈现为横向排列或者环形放射图。这些轨迹的长度通常相等,代表相同的序列区域。可用一个轨迹表示基因的位置,另一个轨迹表示调节区域,第三个轨迹表示GC含量。可将最常用轨迹的特征打包为一个特征集(feature-sets)。CDS的特征可以用一个特征集,而tRNA的特征可以用另外一个特征集。这不是强制性的要求,你可以在diagram中用同样的特征集。如果diagram中用不同的特征集,修改一个特征会很容易,比如把所有tRNA的特征都变为红色,你只需选择tRNA的特征就行。
新建图形主要有两种方式。第一种是自上而下的方法(Top-Down),首先新建diagram对象,然后用diagram的方法来添加track(s),最后用track的方法添加特征。第二种是自下而上的方法(Bottom-Up),首先单独新建对象,然后再将其进行组合。
17.1.3 自上而下的实例¶
我们用一个从GenBank文件中读取出来的 SeqRecord 来绘制全基因组(详见第 5 章)。这里用鼠疫杆菌 Yersinia pestis biovar Microtus 的pPCP1质粒,元数据文件NC_005816.gb在Biopython中GenBank的tests目录下, NC_005816.gb 也可下载
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb", "genbank")
这里用自上而下的方法,导入目标序列后,新建一个diagram,然后新建一个track,最后新建一个特征集(feature set):
gd_diagram = GenomeDiagram.Diagram("Yersinia pestis biovar Microtus plasmid pPCP1")
gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
gd_feature_set = gd_track_for_features.new_set()
接下来的部分最有趣,提取 SeqRecord 中每个基因的 SeqFeature 对象,就会为diagram生成一个相应的特征(feature),将其颜色设置为蓝色,分别用深蓝和浅蓝表示。
for feature in record.features:
if feature.type != "gene":
#Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(feature, color=color, label=True)
创建导出文件需要两步,首先是 draw 方法,它用ReportLab对象生成全部图形。然后是 write 方法,将图形存储到格式文件。注意:输出文件格式不止一种。
gd_diagram.draw(format="linear", orientation="landscape", pagesize='A4',
fragments=4, start=0, end=len(record))
gd_diagram.write("plasmid_linear.pdf", "PDF")
gd_diagram.write("plasmid_linear.eps", "EPS")
gd_diagram.write("plasmid_linear.svg", "SVG")
如果安装了依赖关系(Dependencies),也可以生成位图(Bitmap image),代码如下:
gd_diagram.write("plasmid_linear.png", "PNG")
注意,我们将代码中的 fragments 变量设置为“4”,基因组就会被分为“4”个片段。
如果想要环形图,可以试试以下的代码:
gd_diagram.draw(format="circular", circular=True, pagesize=(20*cm,20*cm),
start=0, end=len(record), circle_core=0.7)
gd_diagram.write("plasmid_circular.pdf", "PDF")
示例图不是非常精彩,但这仅仅是精彩的开始。
17.1.4 自下而上的实例¶
现在,用“自下而上”的方法来创建相同的图形。首先新建不同的对象(可以是任何顺序),然后将其组合。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb", "genbank")
#Create the feature set and its feature objects,
gd_feature_set = GenomeDiagram.FeatureSet()
for feature in record.features:
if feature.type != "gene":
#Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(feature, color=color, label=True)
#(this for loop is the same as in the previous example)
#Create a track, and a diagram
gd_track_for_features = GenomeDiagram.Track(name="Annotated Features")
gd_diagram = GenomeDiagram.Diagram("Yersinia pestis biovar Microtus plasmid pPCP1")
#Now have to glue the bits together...
gd_track_for_features.add_set(gd_feature_set)
gd_diagram.add_track(gd_track_for_features, 1)
同样,利用 draw 和 write 方法来创建线形图或者环形图,结果应该完全相同(“draw”和“write”部分的代码见17.1.3)。
17.1.5 简单的Feature¶
以上示例中,创建diagram使用的 SeqRecord 的 SeqFeature 对象( 详见 4.3 章节)。如果你不需要 SeqFeature 对象,只将目标feature定位在坐标轴,仅需要创建minimal
SeqFeature 对象,方法很简单,代码如下:
from Bio.SeqFeature import SeqFeature, FeatureLocation
my_seq_feature = SeqFeature(FeatureLocation(50,100),strand=+1)
对于序列来说, +1 代表正向, -1 代表反向, None 代表两者都有,下面举个简单的示例:
from Bio.SeqFeature import SeqFeature, FeatureLocation
from Bio.Graphics import GenomeDiagram
from reportlab.lib.units import cm
gdd = GenomeDiagram.Diagram('Test Diagram')
gdt_features = gdd.new_track(1, greytrack=False)
gds_features = gdt_features.new_set()
#Add three features to show the strand options,
feature = SeqFeature(FeatureLocation(25, 125), strand=+1)
gds_features.add_feature(feature, name="Forward", label=True)
feature = SeqFeature(FeatureLocation(150, 250), strand=None)
gds_features.add_feature(feature, name="Strandless", label=True)
feature = SeqFeature(FeatureLocation(275, 375), strand=-1)
gds_features.add_feature(feature, name="Reverse", label=True)
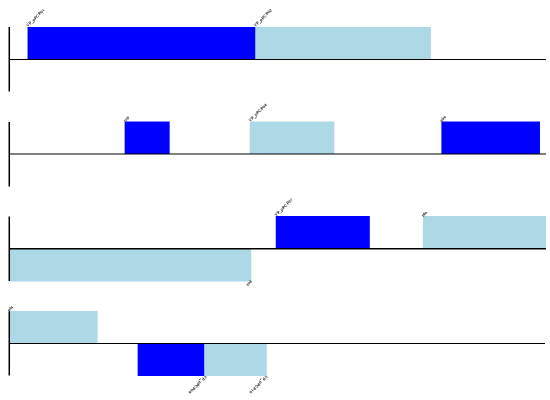
gdd.draw(format='linear', pagesize=(15*cm,4*cm), fragments=1,
start=0, end=400)
gdd.write("GD_labels_default.pdf", "pdf")
图形示例结果请见下一节图中的第一个图,缺省的feature为浅绿色。
注意,这里用 name 参数作为feature的“说明文本”(caption text)。下文将会讲述更多细节。
17.1.6 Feature说明¶
下面代码中, feature 作为 SeqFeature 的对象添加到diagram。
gd_feature_set.add_feature(feature, color=color, label=True)
前面的示例用 SeqFeature 的注释为feature做了恰当的文字说明。 SeqFeature 对象的限定符(qualifiers dictionary)缺省值是: gene, label, name, locus_tag, 和 product 。简单地说,你可以定义一个名称:
gd_feature_set.add_feature(feature, color=color, label=True, name="My Gene")
每个feature标签的说明文本可以设置字体、位置和方向。说明文本默认的位置在图形符号(sigil)的左边,可选择在中间或者右边,线形图中文本的默认方向是45°旋转。
#Large font, parallel with the track
gd_feature_set.add_feature(feature, label=True, color="green",
label_size=25, label_angle=0)
#Very small font, perpendicular to the track (towards it)
gd_feature_set.add_feature(feature, label=True, color="purple",
label_position="end",
label_size=4, label_angle=90)
#Small font, perpendicular to the track (away from it)
gd_feature_set.add_feature(feature, label=True, color="blue",
label_position="middle",
label_size=6, label_angle=-90)
用前面示例的代码将这三个片段组合之后应该可以得到如下的结果:
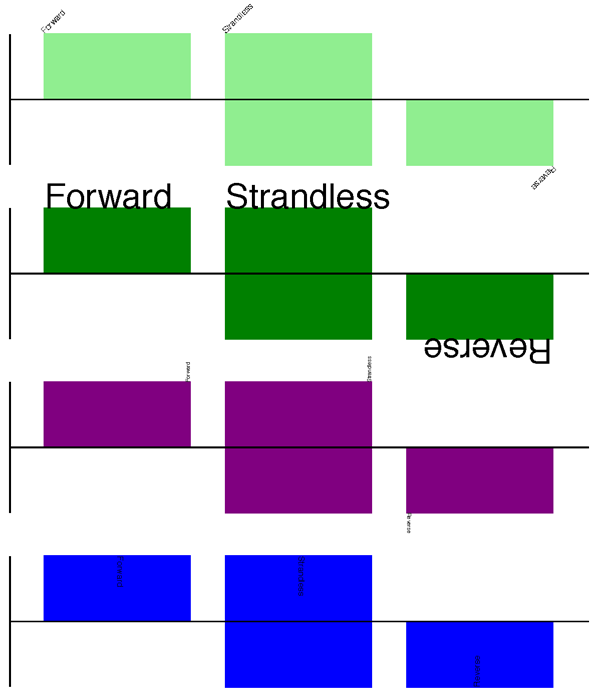
除此之外,还可以设置“label_color”来调节标签的颜色(第 17.1.9 节也将用到这一步),这里没有进行演示。
示例中默认的字体很小,这是比较明智的,因为通常我们会把许多Feature同时展示,而不像这里只展示了几个比较大的feature。
17.1.7 表示Feature的图形符号¶
以上示例中Feature的图形符号(sigil)默认是一个方框(plain box),GenomeDiagram第一版中只有这一选项,后来GenomeDiagram被整合到Biopython1.50时,新增了箭头状的图形符号(sigil)。
#Default uses a BOX sigil
gd_feature_set.add_feature(feature)
#You can make this explicit:
gd_feature_set.add_feature(feature, sigil="BOX")
#Or opt for an arrow:
gd_feature_set.add_feature(feature, sigil="ARROW")
Biopython 1.61又新增3个图形形状(sigil)。
#Box with corners cut off (making it an octagon)
gd_feature_set.add_feature(feature, sigil="OCTO")
#Box with jagged edges (useful for showing breaks in contains)
gd_feature_set.add_feature(feature, sigil="JAGGY")
#Arrow which spans the axis with strand used only for direction
gd_feature_set.add_feature(feature, sigil="BIGARROW")
下面就是这些新增的图形形状(sigil),多数的图形形状都在边界框(bounding box)内部,在坐标轴的上/下位置代表序列(Strand)方向的正/反向,或者上下跨越坐标轴,高度是其他图形形状的两倍。“BIGARROW”有所不同,它总是跨越坐标轴,方向由feature的序列决定。

17.1.8 箭头形状¶
上一部分我们简单引出了箭头形状。还有两个选项可以对箭头形状进行设置:首先根据边界框的高度比例来设置箭杆宽度。
#Full height shafts, giving pointed boxes:
gd_feature_set.add_feature(feature, sigil="ARROW", color="brown",
arrowshaft_height=1.0)
#Or, thin shafts:
gd_feature_set.add_feature(feature, sigil="ARROW", color="teal",
arrowshaft_height=0.2)
#Or, very thin shafts:
gd_feature_set.add_feature(feature, sigil="ARROW", color="darkgreen",
arrowshaft_height=0.1)
结果见下图:
![]()
其次,根据边界框的高度比例设置箭头长度(默认为0.5或50%):
#Short arrow heads:
gd_feature_set.add_feature(feature, sigil="ARROW", color="blue",
arrowhead_length=0.25)
#Or, longer arrow heads:
gd_feature_set.add_feature(feature, sigil="ARROW", color="orange",
arrowhead_length=1)
#Or, very very long arrow heads (i.e. all head, no shaft, so triangles):
gd_feature_set.add_feature(feature, sigil="ARROW", color="red",
arrowhead_length=10000)
结果见下图:
![]()
Biopython1.61新增 BIGARROW 箭头形状,它经常跨越坐标轴,箭头指向”左边“代表”反向“,指向”右边“代表”正向“。
#A large arrow straddling the axis:
gd_feature_set.add_feature(feature, sigil="BIGARROW")
上述 ARROW 形状中的箭杆和箭头设置选项都适用于 BIGARROW 。
17.1.9 完美示例¶
回到”自上而下的示例 Section 17.1.3 中鼠疫杆菌 Yersinia pestis biovar Microtus 的pPCP1质粒,现在使用”图形符号“的高级选项。箭头表示基因,窄框穿越箭头表示限制性内切酶的切割位点。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
from Bio.SeqFeature import SeqFeature, FeatureLocation
record = SeqIO.read("NC_005816.gb", "genbank")
gd_diagram = GenomeDiagram.Diagram(record.id)
gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
gd_feature_set = gd_track_for_features.new_set()
for feature in record.features:
if feature.type != "gene":
#Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(feature, sigil="ARROW",
color=color, label=True,
label_size = 14, label_angle=0)
#I want to include some strandless features, so for an example
#will use EcoRI recognition sites etc.
for site, name, color in [("GAATTC","EcoRI",colors.green),
("CCCGGG","SmaI",colors.orange),
("AAGCTT","HindIII",colors.red),
("GGATCC","BamHI",colors.purple)]:
index = 0
while True:
index = record.seq.find(site, start=index)
if index == -1 : break
feature = SeqFeature(FeatureLocation(index, index+len(site)))
gd_feature_set.add_feature(feature, color=color, name=name,
label=True, label_size = 10,
label_color=color)
index += len(site)
gd_diagram.draw(format="linear", pagesize='A4', fragments=4,
start=0, end=len(record))
gd_diagram.write("plasmid_linear_nice.pdf", "PDF")
gd_diagram.write("plasmid_linear_nice.eps", "EPS")
gd_diagram.write("plasmid_linear_nice.svg", "SVG")
gd_diagram.draw(format="circular", circular=True, pagesize=(20*cm,20*cm),
start=0, end=len(record), circle_core = 0.5)
gd_diagram.write("plasmid_circular_nice.pdf", "PDF")
gd_diagram.write("plasmid_circular_nice.eps", "EPS")
gd_diagram.write("plasmid_circular_nice.svg", "SVG")
输出结果见下图:
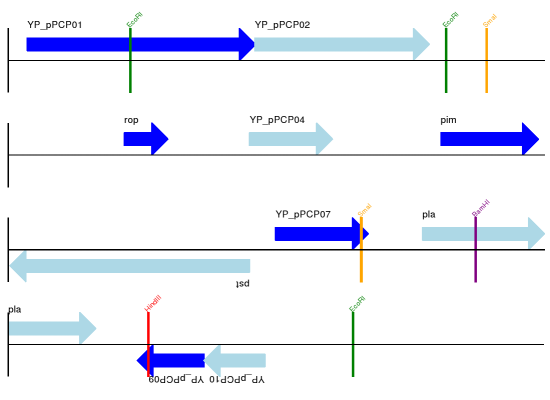
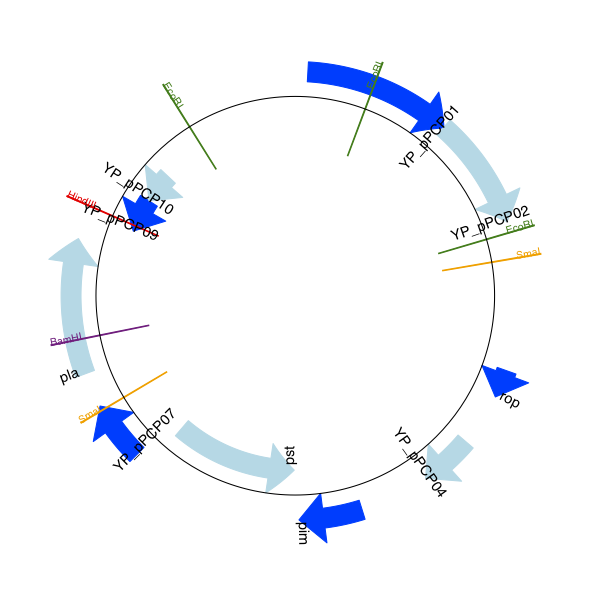
17.1.10 多重轨迹¶
前面实例中都是单独的track,我们可以创建多个track,比如,一个track展示基因,另一个track展示重复序列。Proux等人2002年报道的文章 [5] 中图6是一个很好的范例,下面我们将三个噬菌体基因组依次进行展示。首先需要三个噬菌体的GenBank文件。
NC_002703– Lactococcus phage Tuc2009, 全基因组大小 (38347 bp)AF323668– Bacteriophage bIL285, 全基因组大小(35538 bp)NC_003212– Listeria innocua Clip11262,我们将仅关注前噬菌体5的全基因组 (长度大体相同).
这三个文件可以从Entrez下载,详情请查阅 9.6 。从三个噬菌体基因组文件中分离(slice)提取相关Features信息(请查阅 4.6 ),保证前两个噬菌体的反向互补链与其起始点对齐,再次保存Feature(详情请查阅 4.8 )。
from Bio import SeqIO
A_rec = SeqIO.read("NC_002703.gbk", "gb")
B_rec = SeqIO.read("AF323668.gbk", "gb")
C_rec = SeqIO.read("NC_003212.gbk", "gb")[2587879:2625807].reverse_complement(name=True)
图像中用不同颜色表示基因功能的差异。这需要编辑GenBank文件中每一个feature的颜色参数——就像用 Sanger’s Artemis editor 处理 ——才能被GenomeDiagram识别。但是,这里只需要硬编码(hard code)三个颜色列表。
上述GenBank文件中的注释信息与Proux所用的文件信息并不完全相同,他们还添加了一些未注释的基因。
from reportlab.lib.colors import red, grey, orange, green, brown, blue, lightblue, purple
A_colors = [red]*5 + [grey]*7 + [orange]*2 + [grey]*2 + [orange] + [grey]*11 + [green]*4 \
+ [grey] + [green]*2 + [grey, green] + [brown]*5 + [blue]*4 + [lightblue]*5 \
+ [grey, lightblue] + [purple]*2 + [grey]
B_colors = [red]*6 + [grey]*8 + [orange]*2 + [grey] + [orange] + [grey]*21 + [green]*5 \
+ [grey] + [brown]*4 + [blue]*3 + [lightblue]*3 + [grey]*5 + [purple]*2
C_colors = [grey]*30 + [green]*5 + [brown]*4 + [blue]*2 + [grey, blue] + [lightblue]*2 \
+ [grey]*5
接下来是“draw”方法,给diagram添加3个track。我们在示例中设置不同的开始/结束值来体现它们之间长度不等(Biopython 1.59及更高级的版本)。
from Bio.Graphics import GenomeDiagram
name = "Proux Fig 6"
gd_diagram = GenomeDiagram.Diagram(name)
max_len = 0
for record, gene_colors in zip([A_rec, B_rec, C_rec], [A_colors, B_colors, C_colors]):
max_len = max(max_len, len(record))
gd_track_for_features = gd_diagram.new_track(1,
name=record.name,
greytrack=True,
start=0, end=len(record))
gd_feature_set = gd_track_for_features.new_set()
i = 0
for feature in record.features:
if feature.type != "gene":
#Exclude this feature
continue
gd_feature_set.add_feature(feature, sigil="ARROW",
color=gene_colors[i], label=True,
name = str(i+1),
label_position="start",
label_size = 6, label_angle=0)
i+=1
gd_diagram.draw(format="linear", pagesize='A4', fragments=1,
start=0, end=max_len)
gd_diagram.write(name + ".pdf", "PDF")
gd_diagram.write(name + ".eps", "EPS")
gd_diagram.write(name + ".svg", "SVG")
结果如图所示:
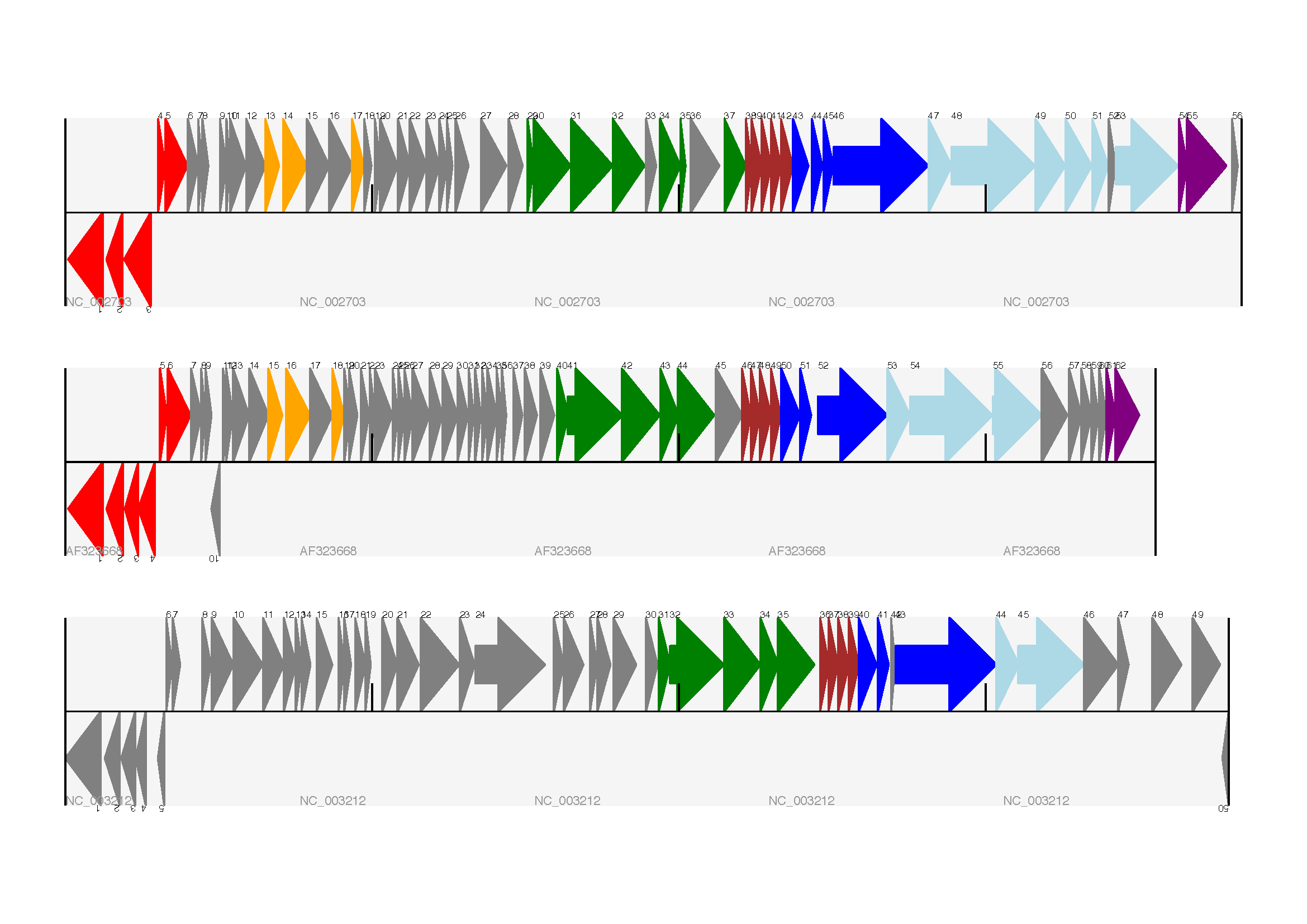
在示例图中底部的噬菌体没有红色或橙色的基因标记。另外,三个噬菌体可视化图的长度不同,这是因为它们的比例相同,长度却不同。
另外有一点不同,不同噬菌体的同源蛋白质之间用有颜色的links相连,下一部分将解决这个问题。
17.1.11 不同Track之间的Cross-Links¶
Biopython 1.59新增绘制不同track之间Cross-Links的功能,这个功能可用于将要展示的简单线形图中,也可用于将线形图分割为短片段(fragments)和环形图。
我们接着模仿Proux等人 [5] 的图像,我们需要一个包含基因之间的“cross links”、“得分”或“颜色”的列表。 实际应用中,可以从BLAST文件自动提取这些信息,这里是手动输入的。
噬菌体的名称同样表示为A,B和C。这里将要展示的是A与B之间的links,噬菌体A和B基因的相似百分比存储在元组中。
#Tuc2009 (NC_002703) vs bIL285 (AF323668)
A_vs_B = [
(99, "Tuc2009_01", "int"),
(33, "Tuc2009_03", "orf4"),
(94, "Tuc2009_05", "orf6"),
(100,"Tuc2009_06", "orf7"),
(97, "Tuc2009_07", "orf8"),
(98, "Tuc2009_08", "orf9"),
(98, "Tuc2009_09", "orf10"),
(100,"Tuc2009_10", "orf12"),
(100,"Tuc2009_11", "orf13"),
(94, "Tuc2009_12", "orf14"),
(87, "Tuc2009_13", "orf15"),
(94, "Tuc2009_14", "orf16"),
(94, "Tuc2009_15", "orf17"),
(88, "Tuc2009_17", "rusA"),
(91, "Tuc2009_18", "orf20"),
(93, "Tuc2009_19", "orf22"),
(71, "Tuc2009_20", "orf23"),
(51, "Tuc2009_22", "orf27"),
(97, "Tuc2009_23", "orf28"),
(88, "Tuc2009_24", "orf29"),
(26, "Tuc2009_26", "orf38"),
(19, "Tuc2009_46", "orf52"),
(77, "Tuc2009_48", "orf54"),
(91, "Tuc2009_49", "orf55"),
(95, "Tuc2009_52", "orf60"),
]
对噬菌体B和C做同样的处理:
#bIL285 (AF323668) vs Listeria innocua prophage 5 (in NC_003212)
B_vs_C = [
(42, "orf39", "lin2581"),
(31, "orf40", "lin2580"),
(49, "orf41", "lin2579"), #terL
(54, "orf42", "lin2578"), #portal
(55, "orf43", "lin2577"), #protease
(33, "orf44", "lin2576"), #mhp
(51, "orf46", "lin2575"),
(33, "orf47", "lin2574"),
(40, "orf48", "lin2573"),
(25, "orf49", "lin2572"),
(50, "orf50", "lin2571"),
(48, "orf51", "lin2570"),
(24, "orf52", "lin2568"),
(30, "orf53", "lin2567"),
(28, "orf54", "lin2566"),
]
噬菌体A和C的标识符(Identifiers)是基因座标签(locus tags),噬菌体B没有基因座标签,这里用基因名称来代替。以下的辅助函数可用基因座标签或基因名称来寻找Feature。
def get_feature(features, id, tags=["locus_tag", "gene"]):
"""Search list of SeqFeature objects for an identifier under the given tags."""
for f in features:
for key in tags:
#tag may not be present in this feature
for x in f.qualifiers.get(key, []):
if x == id:
return f
raise KeyError(id)
现在将这些标识符对(identifier pairs)的列表转换为“SeqFeature”列表,因此来查找它们的坐标定位。现在将下列代码添加到上段代码中 gd_diagram.draw(...) 这一行之前,将cross-links添加到图像中。示例中的脚本文件 Proux_et_al_2002_Figure_6.py 在Biopython源程序文件夹的 Doc/examples 目录下。
from Bio.Graphics.GenomeDiagram import CrossLink
from reportlab.lib import colors
#Note it might have been clearer to assign the track numbers explicitly...
for rec_X, tn_X, rec_Y, tn_Y, X_vs_Y in [(A_rec, 3, B_rec, 2, A_vs_B),
(B_rec, 2, C_rec, 1, B_vs_C)]:
track_X = gd_diagram.tracks[tn_X]
track_Y = gd_diagram.tracks[tn_Y]
for score, id_X, id_Y in X_vs_Y:
feature_X = get_feature(rec_X.features, id_X)
feature_Y = get_feature(rec_Y.features, id_Y)
color = colors.linearlyInterpolatedColor(colors.white, colors.firebrick, 0, 100, score)
link_xy = CrossLink((track_X, feature_X.location.start, feature_X.location.end),
(track_Y, feature_Y.location.start, feature_Y.location.end),
color, colors.lightgrey)
gd_diagram.cross_track_links.append(link_xy)
这段代码有几个要点,第一, GenomeDiagram 对象有一个 cross_track_links 属性,这个属性只是 CrossLink 对象的一组数据。每个 CrossLink 对象有两个track-specific坐标,示例中用元组(tuples)来展现,可用 GenomeDiagram.Feature 对象来代替。可选择添加颜色和边框颜色,还可以说明这个link是否需要翻转,这个功能易于表现染色体异位。
你也可以看我们是如何将BLAST中特征百分比(Percentage Identity Score)转换为白-红的渐变色(白-0%,红-100%)。这个实例中没有cross-links的重叠,如果有links重叠可以用ReportLab库中的透明度(transparency)来解决,通过设置颜色的alpha通道来使用。然而,若同时使用边框阴影和叠加透明度会增加理解的难度。结果见下图:
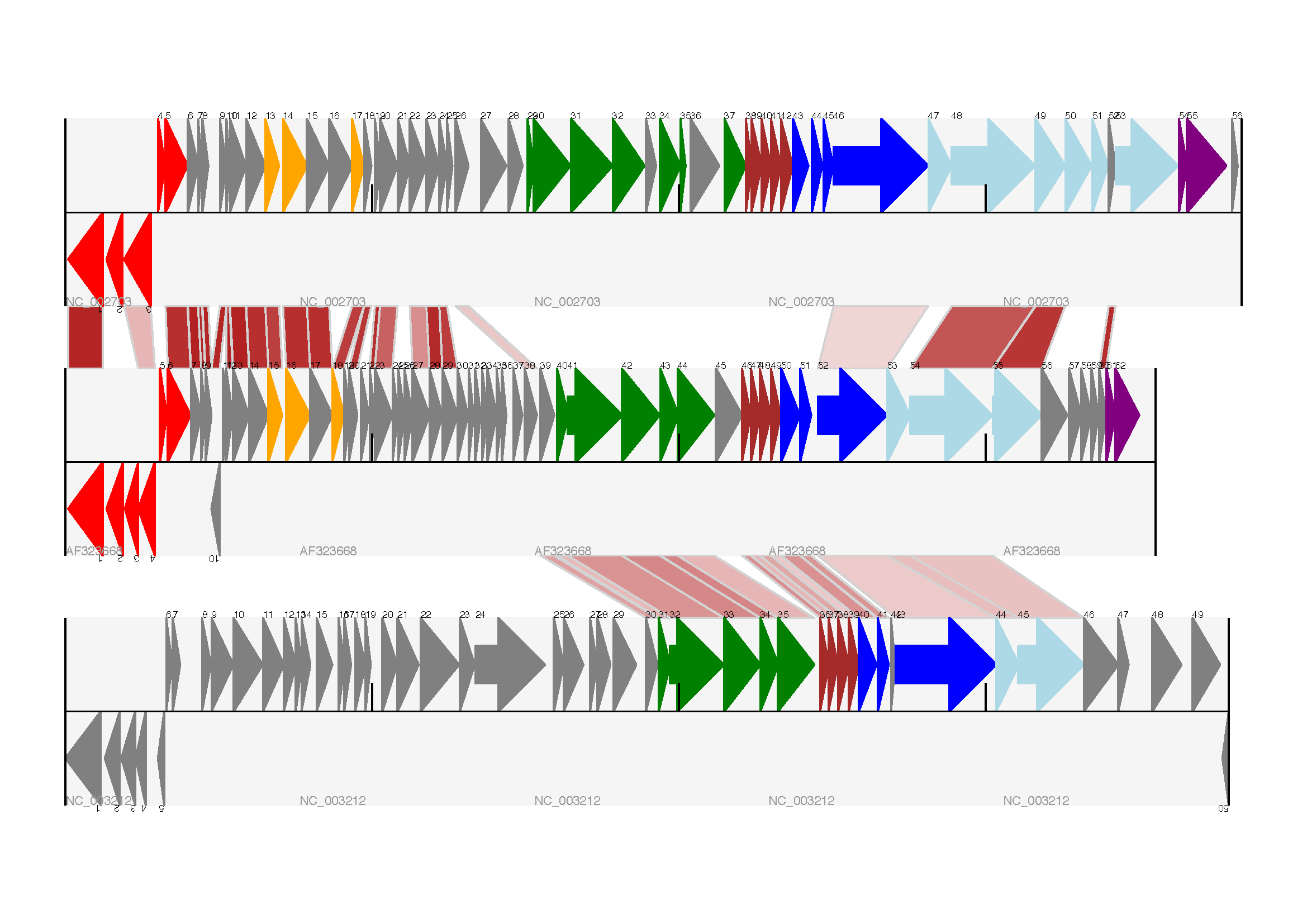
当然,Biopython还有很多增强图像效果的方法。首先,这个示例中的cross links是蛋白质之间的,被呈现在一个链的固定区域(strand specific manor)。可以在feature track上用 ‘BOX’ sigil添加背景区域(background region)来扩展cross link的效果。同样,可以缩短feature tracks之间的垂直高度,使用更多的links来代替——一种方法是为空的track分配空间。此外,在没有大规模基因重叠的情况下,可以用跨越轴线的”BIGARROW”,这样就为track进一步增加了垂直空间。详情请查看Biopython源程序的 Doc/examples 目录下的示例脚本文件:Proux_et_al_2002_Figure_6.py 。
结果见下图:
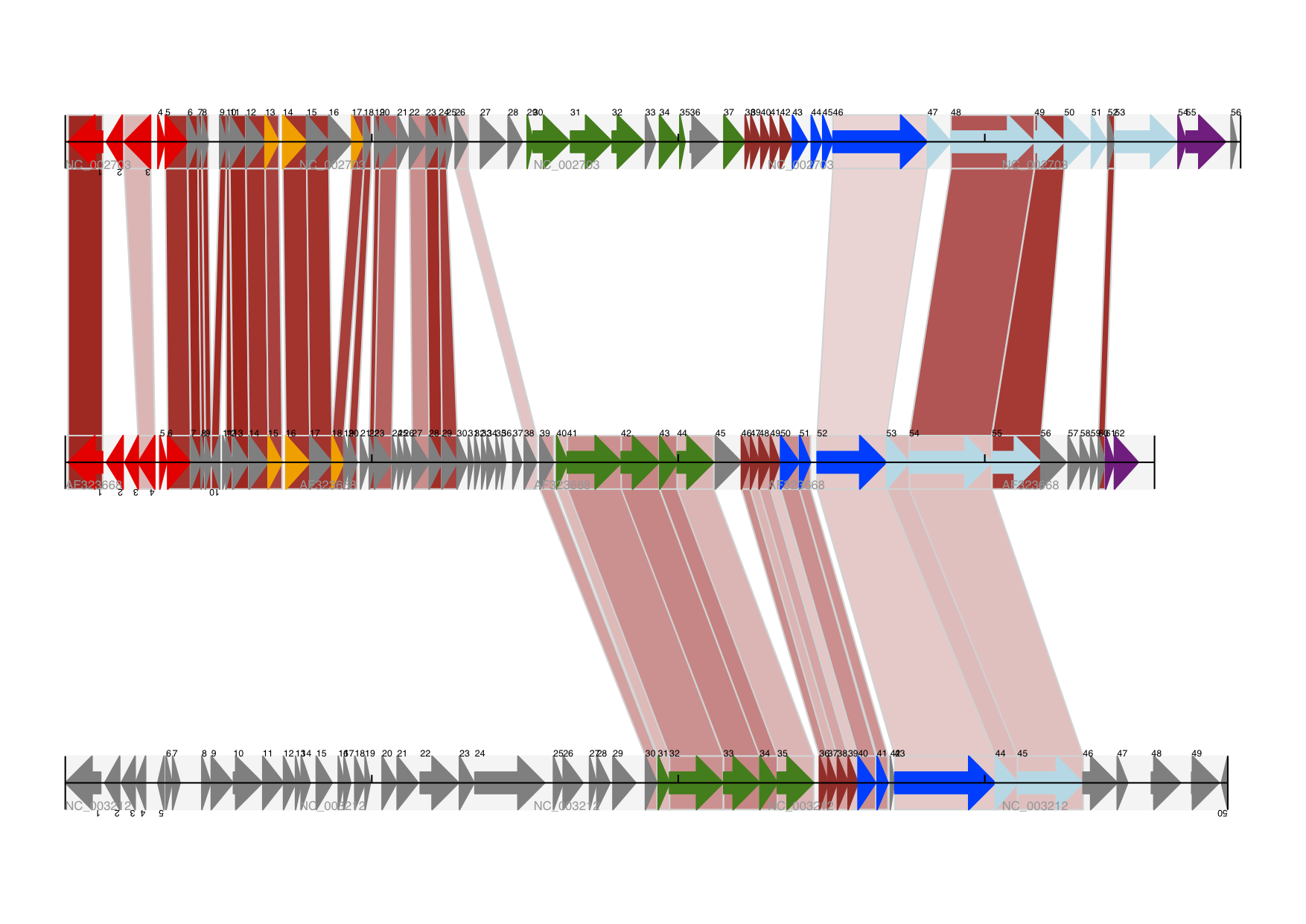
除此之外,你可能希望在图像编辑软件里手动调整gene标签的位置,添加特定标识,比如强调某个特别的区域。
如果有多个叠加的links,使用ReportLab库里的颜色透明度(transparent color)是非常好的方法,由于这个示例没有cross-link的重叠,所以没有用到颜色透明度(transparent color)。然而,尽量避免在这个示例中使用边框阴影(shaded color scheme)。
17.1.12 高级选项¶
可以通过控制刻度线(tick marks)来调节展示比例(scale),毕竟每个图形应该包括基本单位和轴线标签的数目。
到目前为止,我们只使用了 FeatureSet 。GenomeDiagram还可以用 GraphSet 来制作线形图,饼状图和heatmap热图(例如在轨迹内展示feature中的GC含量)。
目前还没有添加这个选项,最后,推荐你去参考GenomeDiagram单机版 用户指南 (PDF) 和文档字符串(docstrings)。
17.1.13 转换旧代码¶
如果你有用GenomeDiagram独立版本写的旧代码,想将其转换为Bippython和新版本可识别的代码,你需要做一些调整——主要是import语句。GenomeDiagram的旧版本中使用英式拼写“colour” 和 “centre”来表示“color” 和“center”。被Biopython整合后,参数名可以使用任意一种。但是将来可能会不支持英式的参数名。
如果你过去使用下面的方式:
from GenomeDiagram import GDFeatureSet, GDDiagram
gdd = GDDiagram("An example")
...
你只需要将import语句转换成下面这样:
from Bio.Graphics.GenomeDiagram import FeatureSet as GDFeatureSet, Diagram as GDDiagram
gdd = GDDiagram("An example")
...
希望能够顺利运行。将来你可能想换用新名称,你必须在更大程度上改变你编写代码的方式:
from Bio.Graphics.GenomeDiagram import FeatureSet, Diagram
gdd = Diagram("An example")
...
or:
from Bio.Graphics import GenomeDiagram
gdd = GenomeDiagram.Diagram("An example")
...
如果运行过程中出现问题,请到Biopython邮件列表中寻求帮助。唯一的缺点就是没有包括旧模块 GenomeDiagram.GDUtilities ,这个模块有计算GC百分比含量的函数,这一部分将会合并到 Bio.SeqUtils 模块。
17.2 染色体¶
Bio.Graphics.BasicChromosome 模块可以绘制染色体,Jupe等人在2012发表的文章 [6] 中利用不同的颜色来展示不同的基因家族。
17.2.1 简单染色体¶
我们用 Arabidopsis thaliana 来展示一个简单示例。
首先从NCBI的FTP服务器 ftp://ftp.ncbi.nlm.nih.gov/genomes/Arabidopsis_thaliana 下载拟南芥已测序的五个染色体文件,利用 Bio.SeqIO 函数计算它们的长度。你可以利用GenBank文件,但是对于染色体来说,FASTA文件的处理速度会快点。
from Bio import SeqIO
entries = [("Chr I", "CHR_I/NC_003070.fna"),
("Chr II", "CHR_II/NC_003071.fna"),
("Chr III", "CHR_III/NC_003074.fna"),
("Chr IV", "CHR_IV/NC_003075.fna"),
("Chr V", "CHR_V/NC_003076.fna")]
for (name, filename) in entries:
record = SeqIO.read(filename,"fasta")
print name, len(record)
计算出5个染色体长度后,就可用 BasicChromosome 模块对其作如下的处理:
from reportlab.lib.units import cm
from Bio.Graphics import BasicChromosome
entries = [("Chr I", 30432563),
("Chr II", 19705359),
("Chr III", 23470805),
("Chr IV", 18585042),
("Chr V", 26992728)]
max_len = 30432563 #Could compute this
telomere_length = 1000000 #For illustration
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7*cm, 21*cm) #A4 landscape
for name, length in entries:
cur_chromosome = BasicChromosome.Chromosome(name)
#Set the scale to the MAXIMUM length plus the two telomeres in bp,
#want the same scale used on all five chromosomes so they can be
#compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length
#Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
#Add a body - using bp as the scale length here.
body = BasicChromosome.ChromosomeSegment()
body.scale = length
cur_chromosome.add(body)
#Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
#This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("simple_chrom.pdf", "Arabidopsis thaliana")
新建的PDF文档如图所示:
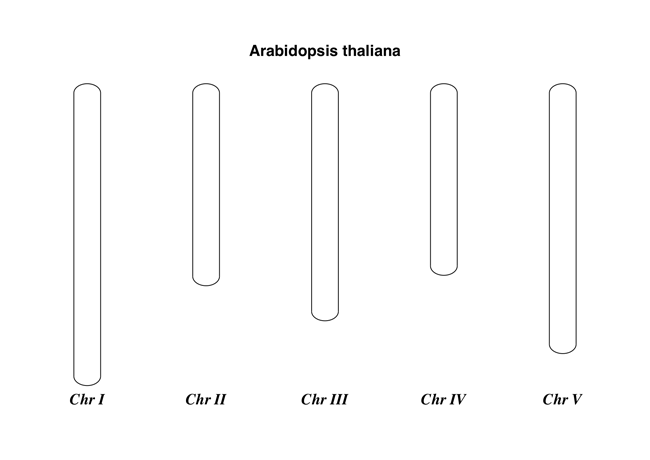
这个示例可以短小精悍,下面的示例可以展示目标feature的定位。
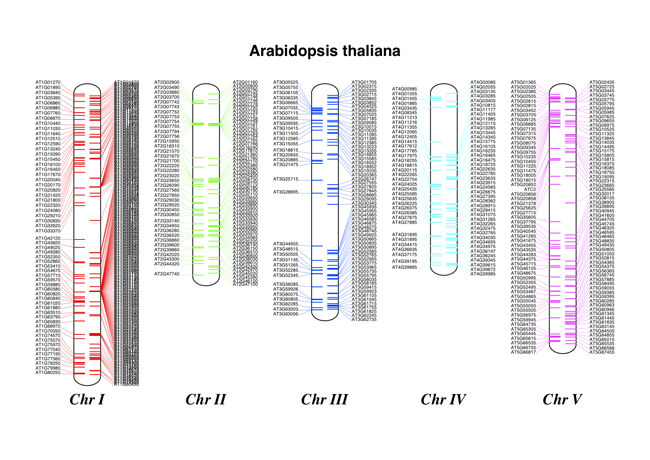
17.2.2 染色体注释¶
继续前面的示例,我们可以同时展示tRNA基因。通过解析 Arabidopsis thaliana 的5个染色体GenBank文件,我们可以对他们进行定位。你需要从NCBI的FTP服务器下载这些文件 ftp://ftp.ncbi.nlm.nih.gov/genomes/Arabidopsis_thaliana ,也可以保存子目录名称或者添加如下的路径:
from reportlab.lib.units import cm
from Bio import SeqIO
from Bio.Graphics import BasicChromosome
entries = [("Chr I", "CHR_I/NC_003070.gbk"),
("Chr II", "CHR_II/NC_003071.gbk"),
("Chr III", "CHR_III/NC_003074.gbk"),
("Chr IV", "CHR_IV/NC_003075.gbk"),
("Chr V", "CHR_V/NC_003076.gbk")]
max_len = 30432563 #Could compute this
telomere_length = 1000000 #For illustration
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7*cm, 21*cm) #A4 landscape
for index, (name, filename) in enumerate(entries):
record = SeqIO.read(filename,"genbank")
length = len(record)
features = [f for f in record.features if f.type=="tRNA"]
#Record an Artemis style integer color in the feature's qualifiers,
#1 = Black, 2 = Red, 3 = Green, 4 = blue, 5 =cyan, 6 = purple
for f in features: f.qualifiers["color"] = [index+2]
cur_chromosome = BasicChromosome.Chromosome(name)
#Set the scale to the MAXIMUM length plus the two telomeres in bp,
#want the same scale used on all five chromosomes so they can be
#compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length
#Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
#Add a body - again using bp as the scale length here.
body = BasicChromosome.AnnotatedChromosomeSegment(length, features)
body.scale = length
cur_chromosome.add(body)
#Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
#This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("tRNA_chrom.pdf", "Arabidopsis thaliana")
如果标签之间太紧密会发出警告,所以要注意第一条染色体的的前导链(左手边),可以创建一个彩色的PDF文件,如下图所示: