第18章 Cookbook – 用它做一些很酷的事情¶
Biopython目前有两个版本的“cookbook”示例——本章(本章包含在教程中许多年,并渐渐成熟),和在Biopython维基上的由用户贡献的集合: http://biopython.org/wiki/Category:Cookbook 。
我们在试着鼓励Biopython用户在维基上贡献他们自己的示例。除了能帮助社区之外,分享像这样的示例的一个直接的好处是,你也能从其他Biopython用户和开发者中获得一些关于代码的意见反馈——这或许能帮助你改进自己的Python代码。
长期来说,我们可能最终会将这一章所有的示例都转移到维基上,或者本教程其他的地方。
18.1 操作序列文件¶
这部分将展示更多使用第 5 章所描述的 Bio.SeqIO 模块来进行序列输入/输出操作的例子。
18.1.1 过滤文件中的序列¶
通常你会拥有一个包含许多序列的大文件(例如,FASTA基因文件,或者FASTQ或SFF读长文件),和一个包含你所感兴趣的序列的ID列表,而你希望创建一个由这一ID列表里的序列构成的文件。
让我们假设这个ID列表在一个简单的文本文件中,作为每一行的第一个词。这可能是一个表格文件,其第一列是序列ID。尝试下面的代码:
from Bio import SeqIO
input_file = "big_file.sff"
id_file = "short_list.txt"
output_file = "short_list.sff"
wanted = set(line.rstrip("\n").split(None,1)[0] for line in open(id_file))
print "Found %i unique identifiers in %s" % (len(wanted), id_file)
records = (r for r in SeqIO.parse(input_file, "sff") if r.id in wanted)
count = SeqIO.write(records, output_file, "sff")
print "Saved %i records from %s to %s" % (count, input_file, output_file)
if count < len(wanted):
print "Warning %i IDs not found in %s" % (len(wanted)-count, input_file)
注意,我们使用Python的 set 类型而不是 list,这会使得检测成员关系更快。
18.1.2 生成随机基因组¶
假设你在检视一个基因组序列,寻找一些序列特征——或许是极端局部GC含量偏差,或者可能的限制性酶切位点。一旦你使你的Python代码在真实的基因组上运行后,尝试在相同的随机化版本基因组上运行,并进行统计分析或许是明智的选择(毕竟,任何你发现的“特性”都可能只是偶然事件)。
在这一讨论中,我们将使用来自 Yersinia pestis biovar Microtus 的pPCP1质粒的GenBank文件。该文件包含在Biopython单元测试的GenBank文件夹中,或者你可以从我们的网站上得到, NC_005816.gb. 该文件仅有一个记录,所以我们能用 Bio.SeqIO.read() 函数把它当做 SeqRecord 读入:
>>> from Bio import SeqIO
>>> original_rec = SeqIO.read("NC_005816.gb","genbank")
那么,我们怎么生成一个原始序列重排后的版本能?我会使用Python内置的 random 模块来做这个,特别是 random.shuffle 函数——但是这个只作用于Python列表。我们的序列是一个 Seq 对象,所以为了重排它,我们需要将它转换为一个列表:
>>> import random
>>> nuc_list = list(original_rec.seq)
>>> random.shuffle(nuc_list) #acts in situ!
现在,为了使用 Bio.SeqIO 输出重排的序列,我们需要使用重排后的列表重新创建包含一个新的 SeqRecord 包含随即化后的 Seq 。要实现这个,我们需要将核苷酸(单字母字符串)列表转换为长字符串——在Python中,一般使用字符串对象的join方法来实现它。
>>> from Bio.Seq import Seq
>>> from Bio.SeqRecord import SeqRecord
>>> shuffled_rec = SeqRecord(Seq("".join(nuc_list), original_rec.seq.alphabet),
... id="Shuffled", description="Based on %s" % original_rec.id)
让我们将所有这些片段放到一起来组成一个完整的Python脚本,这个脚本将生成一个FASTA序列文件,其包含30个原始序列的随机重排版本。
第一个版本只是使用一个大的for循环,并一个一个的输出记录(使用章节 5.5.4 描述的``SeqRecord`` 的格式化方法):
import random
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio import SeqIO
original_rec = SeqIO.read("NC_005816.gb","genbank")
handle = open("shuffled.fasta", "w")
for i in range(30):
nuc_list = list(original_rec.seq)
random.shuffle(nuc_list)
shuffled_rec = SeqRecord(Seq("".join(nuc_list), original_rec.seq.alphabet), \
id="Shuffled%i" % (i+1), \
description="Based on %s" % original_rec.id)
handle.write(shuffled_rec.format("fasta"))
handle.close()
我个人更喜欢下面的版本(不使用for循环),而使用一个函数来重排记录以及一个生成表达式:
import random
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio import SeqIO
def make_shuffle_record(record, new_id):
nuc_list = list(record.seq)
random.shuffle(nuc_list)
return SeqRecord(Seq("".join(nuc_list), record.seq.alphabet), \
id=new_id, description="Based on %s" % original_rec.id)
original_rec = SeqIO.read("NC_005816.gb","genbank")
shuffled_recs = (make_shuffle_record(original_rec, "Shuffled%i" % (i+1)) \
for i in range(30))
handle = open("shuffled.fasta", "w")
SeqIO.write(shuffled_recs, handle, "fasta")
handle.close()
18.1.3 翻译CDS条目为FASTA文件¶
假设你有一个包含某个物种的CDS条目作为输入文件,你想生成一个由它们的蛋白序列组成的FASTA文件。也就是,从原始文件中取出每一个核苷酸序列,并翻译它。回到章节 3.9 我们了解了怎么使用 Seq 对象的 translate 方法,和可选的 cds 参数来使得不同的起始密码子能正确翻译。
就像章节 5.5.3 中反向互补的例子中展示的那样,我们可以用 Bio.SeqIO 将与翻译步骤结合起来。对于每一个核苷酸 SeqRecord ,我们需要创建一个蛋白的 SeqRecord —— 并对它命名。
你能编写自己的函数来做这个事情,为你的序列选择合适的蛋白标识和恰当的密码表。在本例中,我们仅使用默认的密码表,并给序列ID加一个前缀。
from Bio.SeqRecord import SeqRecord
def make_protein_record(nuc_record):
"""Returns a new SeqRecord with the translated sequence (default table)."""
return SeqRecord(seq = nuc_record.seq.translate(cds=True), \
id = "trans_" + nuc_record.id, \
description = "translation of CDS, using default table")
我们能用这个函数将核苷酸记录转换为蛋白记录,作为输出。一个优雅且内存高效的方式是使用生成表达式(generator expression):
from Bio import SeqIO
proteins = (make_protein_record(nuc_rec) for nuc_rec in \
SeqIO.parse("coding_sequences.fasta", "fasta"))
SeqIO.write(proteins, "translations.fasta", "fasta")
以上代码适用于完整编码序列的FASTA文件。如果你使用部分编码序列,你可能希望在以上的例子中使用 nuc_record.seq.translate(to_stop=True) ,这会告诉Biopython不检查起始密码的有效性,等等。
18.1.4 将FASTA文件中的序列变为大写¶
通常你会从合作者那里得到FASTA文件的数据,有时候这些序列可能是大小写混合的。在某些情况下,这些可能是有意为之的(例如,小写的作为低质量的区域),但通常大小写并不重要。你可能希望编辑这个文件以使所有的序列都变得一致(如,都为大写),你可以使用 SeqRecord 对象的 upper() 方法轻易的实现(Biopython 1.55中引入):
from Bio import SeqIO
records = (rec.upper() for rec in SeqIO.parse("mixed.fas", "fasta"))
count = SeqIO.write(records, "upper.fas", "fasta")
print "Converted %i records to upper case" % count
这是怎么工作的呢?第一行只是导入 Bio.SeqIO 模块。第二行是最有趣的——这是一个Python生成器表达式,它提供 mixed.fas 里每个记录的大写版本。第三行中,我们把这个生成器表达式传给 Bio.SeqIO.write() 函数,它会把大写的序列写出到 upper.fas 输出文件。
我们使用生成器(而不是一个列表或列表解析式)的原因是,前一方式每次仅有一个记录保存在内存中。当你在处理包含成千上万的条目的文件时,这可能非常重要。
18.1.5 对序列文件排序¶
假设你想对一个序列文件按序列长度排序(例如,一个序列拼接的重叠群(contig)集合),而你工作的文件格式可能是像FASTA或FASTQ这样 Bio.SeqIO 能读写(和索引)的格式。
如果文件足够小,你能将它都一次读入内存为一个 SeqRecord 对象列表,对列表进行排序,并保存它:
from Bio import SeqIO
records = list(SeqIO.parse("ls_orchid.fasta","fasta"))
records.sort(cmp=lambda x,y: cmp(len(x),len(y)))
SeqIO.write(records, "sorted_orchids.fasta", "fasta")
唯一巧妙的地方是指明一个比较函数来说明怎样对记录进行排序(这里我们按长度对他们排序)。如果你希望最长的记录在第一个,你可以交换比对,或者使用reverse参数:
from Bio import SeqIO
records = list(SeqIO.parse("ls_orchid.fasta","fasta"))
records.sort(cmp=lambda x,y: cmp(len(y),len(x)))
SeqIO.write(records, "sorted_orchids.fasta", "fasta")
现在这一实现是非常直接的——但是如果你的文件非常大,你不能像这样把它整个加载到内存中应该怎么办呢?例如,你可能有一些二代测序的读长要根据长度排序。这时你可以使用 Bio.SeqIO.index() 函数解决。
from Bio import SeqIO
#Get the lengths and ids, and sort on length
len_and_ids = sorted((len(rec), rec.id) for rec in \
SeqIO.parse("ls_orchid.fasta","fasta"))
ids = reversed([id for (length, id) in len_and_ids])
del len_and_ids #free this memory
record_index = SeqIO.index("ls_orchid.fasta", "fasta")
records = (record_index[id] for id in ids)
SeqIO.write(records, "sorted.fasta", "fasta")
首先我们使用 Bio.SeqIO.parse() 来将整个文件扫描一遍,并将所有记录的标识和它们的长度保存在一个元组(tuple)中。接着我们对这个元组按照序列长度进行排序,并舍弃这些长度。有了这一排列后的标识列表, Bio.SeqIO.index() 允许我们一个一个获取这些记录,我们把它们传给 Bio.SeqIO.write() 输出。
这些例子都使用 Bio.SeqIO 来解析记录为 SeqRecord 对象,并通过 Bio.SeqIO.write() 输出。当你想排序的文件格式 Bio.SeqIO.write() 不支持应该怎么办呢?如纯文本的SwissProt格式。这里有一个额外的解决方法——使用在 Biopython 1.54 (见 5.4.2.2 )中 Bio.SeqIO.index() 添加的 get_raw() 方法。
from Bio import SeqIO
#Get the lengths and ids, and sort on length
len_and_ids = sorted((len(rec), rec.id) for rec in \
SeqIO.parse("ls_orchid.fasta","fasta"))
ids = reversed([id for (length, id) in len_and_ids])
del len_and_ids #free this memory
record_index = SeqIO.index("ls_orchid.fasta", "fasta")
handle = open("sorted.fasta", "w")
for id in ids:
handle.write(record_index.get_raw(id))
handle.close()
作为一个奖励,由于以上例子不重复将数据解析为 SeqRecord 对象,所以它会更快。
18.1.6 FASTQ文件的简单质量过滤¶
FASTQ文件格式在Sanger被引入,目前被广泛用来存储核苷酸序列(reads)和它们的测序质量。FASTQ文件(和相关的QUAL文件)是单字母注释(per-letter-annotation)的最好的例子,因为序列中每一个核苷酸都有一个相对应的质量分数。任何单字母注释都以list、tuple或string被保存在 SeqRecord 的 letter_annotations 字典中(单字符注释具有和序列长度相同个数的元素)。
一个常见的工作是输入一个大的测序读长集合,并根据它们的质量分数过滤它们(或修剪它们)。下面的例子非常简单,然而足以展示处理 SeqRecord 对象中质量数据的基本用法。我们所有要做的事情是读入一个FASTQ文件,过滤并取出那些PHRED质量分数在某个阈值(这里为20)以上的序列。
在这个例子中,我们将使用从ENA序列读长存档下载的真实数据, ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz (2MB) ,解压后为19MB的文件 SRR020192.fastq 。这是在Roche 454 GS FLX测序平台生成的感染加利福利亚海狮的病毒单端数据(参见 http://www.ebi.ac.uk/ena/data/view/SRS004476 )。
首先,让我们来统计reads的数目:
from Bio import SeqIO
count = 0
for rec in SeqIO.parse("SRR020192.fastq", "fastq"):
count += 1
print "%i reads" % count
现在让我们做一个简单的过滤,PHRED质量不小于20:
from Bio import SeqIO
good_reads = (rec for rec in \
SeqIO.parse("SRR020192.fastq", "fastq") \
if min(rec.letter_annotations["phred_quality"]) >= 20)
count = SeqIO.write(good_reads, "good_quality.fastq", "fastq")
print "Saved %i reads" % count
这只取出了41892条读长中的14580条。一个更有意义的做法是根据质量来裁剪reads,但是这里只是作为一个例子。
FASTQ文件可以包含上百万的记录,所以最好避免一次全部加载它们到内存。这个例子使用一个生成器表达式,这意味着每次只有内存里只有一个 SeqRecord 被创建 —— 避免内存限制。
18.1.7 切除引物序列¶
在这个例子中,假设我们需要寻找一个FASTQ数据中以 GATGACGGTGT 为5’端的引物序列的reads。同上面的例子一样,我们将使用从ENA下载的 SRR020192.fastq 文件( ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz )。该方式同样适用于任何其他Biopython支持的格式(例如FASTA文件)。
这个代码使用 Bio.SeqIO 和一个生成器表达式(避免一次加载所有的序列到内存中),以及 Seq 对象的 startswith 方法来检查读长是否以引物序列开始:
from Bio import SeqIO
primer_reads = (rec for rec in \
SeqIO.parse("SRR020192.fastq", "fastq") \
if rec.seq.startswith("GATGACGGTGT"))
count = SeqIO.write(primer_reads, "with_primer.fastq", "fastq")
print "Saved %i reads" % count
这将从 SRR014849.fastq 找到13819条读长记录,并保存为一个新的FASTQ文件——with_primer.fastq。
现在,假设你希望创建一个包含这些读长,但去除了所有引物序列的FASTQ文件。只需要很小的修改,我们就能对 SeqRecord 进行切片(参见章节 4.6 )以移除前11个字母(我们的引物长度):
from Bio import SeqIO
trimmed_primer_reads = (rec[11:] for rec in \
SeqIO.parse("SRR020192.fastq", "fastq") \
if rec.seq.startswith("GATGACGGTGT"))
count = SeqIO.write(trimmed_primer_reads, "with_primer_trimmed.fastq", "fastq")
print "Saved %i reads" % count
这也将从 SRR020192.fastq 取出13819条读长,但是移除了前十个字符,并将它们保存为另一个新的FASTQ文件, with_primer_trimmed.fastq 。
最后,假设你想移除部分reads中的引物并创建一个新的FASTQ文件,而其他的reads保持不变。如果我们仍然希望使用生成器表达式,声明我们自己的修剪(trim)函数可能更加清楚:
from Bio import SeqIO
def trim_primer(record, primer):
if record.seq.startswith(primer):
return record[len(primer):]
else:
return record
trimmed_reads = (trim_primer(record, "GATGACGGTGT") for record in \
SeqIO.parse("SRR020192.fastq", "fastq"))
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print "Saved %i reads" % count
以上代码会运行较长的时间,因为这次输出文件包含所有41892个reads。再次,我们将使用生成器表达式来避免内存问题。你也可以使用一个生成器函数来替代生成器表达式。
from Bio import SeqIO
def trim_primers(records, primer):
"""Removes perfect primer sequences at start of reads.
This is a generator function, the records argument should
be a list or iterator returning SeqRecord objects.
"""
len_primer = len(primer) #cache this for later
for record in records:
if record.seq.startswith(primer):
yield record[len_primer:]
else:
yield record
original_reads = SeqIO.parse("SRR020192.fastq", "fastq")
trimmed_reads = trim_primers(original_reads, "GATGACGGTGT")
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print "Saved %i reads" % count
这种形式非常灵活,如果你想做一些更复杂的事情,譬如只保留部分记录 —— 像下一个例子中展示的那样。
18.1.8 切除接头序列¶
这实际上是前面例子的一个简单扩展。我们将假设 GATGACGGTGT 是某个FASTQ格式数据的一个接头序列,并再次使用来自NCBI的 SRR020192.fastq 文件 ( ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz )。
然而在本例中,我们将在读长的 任何位置 查找序列,不仅仅是最开始:
from Bio import SeqIO
def trim_adaptors(records, adaptor):
"""Trims perfect adaptor sequences.
This is a generator function, the records argument should
be a list or iterator returning SeqRecord objects.
"""
len_adaptor = len(adaptor) #cache this for later
for record in records:
index = record.seq.find(adaptor)
if index == -1:
#adaptor not found, so won't trim
yield record
else:
#trim off the adaptor
yield record[index+len_adaptor:]
original_reads = SeqIO.parse("SRR020192.fastq", "fastq")
trimmed_reads = trim_adaptors(original_reads, "GATGACGGTGT")
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print "Saved %i reads" % count
因为我们在这个例子中使用的是FASTQ文件, SeqRecord 对象包括reads质量分数的单字母注释(per-letter-annotation)。我们可以通过对具有一定质量分数的 SeqRecord 对象进行切片,并将返回的结果保存到一个FASTQ文件。
和上面的例子(只在每个读长的开始查找引物/接头)相比,你会发现有些reads剪切后非常短(例如,如果接头序列在读长的中部发现,而不是开始附近)。所以,让我们再加入一个最低长度要求:
from Bio import SeqIO
def trim_adaptors(records, adaptor, min_len):
"""Trims perfect adaptor sequences, checks read length.
This is a generator function, the records argument should
be a list or iterator returning SeqRecord objects.
"""
len_adaptor = len(adaptor) #cache this for later
for record in records:
len_record = len(record) #cache this for later
if len(record) < min_len:
#Too short to keep
continue
index = record.seq.find(adaptor)
if index == -1:
#adaptor not found, so won't trim
yield record
elif len_record - index - len_adaptor >= min_len:
#after trimming this will still be long enough
yield record[index+len_adaptor:]
original_reads = SeqIO.parse("SRR020192.fastq", "fastq")
trimmed_reads = trim_adaptors(original_reads, "GATGACGGTGT", 100)
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print "Saved %i reads" % count
通过改变格式名称,你也可以将这个应用于FASTA文件。该代码也可以扩展为模糊匹配,而非绝对匹配(或许用一个两两比对,或者考虑读长的质量分数),但是这会使代码变得更慢。
18.1.9 转换FASTQ文件¶
回到章节 5.5.2 ,我们展示了怎样使用 Bio.SeqIO 来实现两个文件格式间的转换。这里,我们将更进一步探讨二代DNA测序中使用的FASTQ文件。更加详细的介绍可以参加 Cock et al. (2009) [7] 。FASTQ文件同时存储DNA序列(以Python字符串的形式)和相应的读长质量。
PHRED分数(在大多数FASTQ文件中使用,也存在于QUAL、ACE和SFF文件中)已经成为一个用来表示某个给定碱基测序错误概率(这里用 P*e* 表示)的 实际 标准(使用一个以10为底的对数转换):
这意味着一个错误的读长( P*e* = 1 )得到的PHRED质量为0,而一个非常好的 P*e* = 0.00001 的读长得到的PHRED质量为50。在实际的测序数据中,质量比这个要高的非常稀少,通过后期处理,如读长映射(mapping)和组装,PHRED质量到达90是可能的(确实,MAQ工具允许PHRED分数在0到93之间)。
FASTQ格式有潜力成为以单文件纯文本方式存储测序读长的字符和质量分数的 实际 的标准。 唯一的美中不足是,目前至少有三个FASTQ格式版本,它们相互并不兼容,且难以区分...
- 原始的Sanger FASTQ格式将PHRED质量分数和33个ASCII字符偏移进行编码。NCBI目前在它们的Short Read Archive中使用这种格式。我们在
Bio.SeqIO中称之为fastq(或fastq-sanger)格式。 - Solexa(后来由Illumina收购)引入了他们自己的版本,使用Solexa质量分数和64个ASCII字符偏移进行编码。我们叫做
fastq-solexa格式。 - Illumina工作流1.3进一步推出了PHRED质量分数(更为一致的版本)的FASTQ文件,但是却以64个ASCII字符偏移编码。我们叫做
fastq-illumina格式。
Solexa质量分数采用一种不同的对数转换:
由于Solexa/Illumina目前在他们的1.3版本的工作流程中已迁移到使用PHRED分数,Solexa质量分数将逐渐淡出使用。如果你将错误估值取等号( P*e* ),这两个等式允许在两个评分系统之间进行转换 —— Biopython在 Bio.SeqIO.QualityIO 模块中有函数可以实现。这一模块在使用 Bio.SeqIO 进行从Solexa/Illumina老文件格式到标准Sanger FASTQ文件格式转换时被调用:
from Bio import SeqIO
SeqIO.convert("solexa.fastq", "fastq-solexa", "standard.fastq", "fastq")
如果你想转换新的Illumina 1.3+ FASTQ文件,改变只会导致ASCII码的整体偏移。因为尽管编码不同,所有的质量分数都是PHRED分数:
from Bio import SeqIO
SeqIO.convert("illumina.fastq", "fastq-illumina", "standard.fastq", "fastq")
注意,像这样使用 Bio.SeqIO.convert() 会比 Bio.SeqIO.parse() 和 Bio.SeqIO.write() 组合快得 多 ,因为转换FASTQ(包括FASTQ到FASTA的转换)的代码经过优化。
对于质量好的读长,PHRED和Solexa分数几乎相等,这意味着,因为 fasta-solexa 和 fastq-illumina 都使用64个ASCII字符偏移,它们的文件几乎相同。这是Illumina有意设计的,也意味着使用老版本 fasta-solexa 格式文件的应用或许也能接受新版本 fastq-illumina 格式文件(在高质量的数据上)。当然,两个版本和原始的,由Sanger、NCBI和其他地方使用的FASTQ标准有很大不同(格式名为 fastq 或 fastq-sanger )。
了解更多细节,请参见内置的帮助(或 在线帮助 ):
>>> from Bio.SeqIO import QualityIO
>>> help(QualityIO)
...
18.1.10 转换FASTA和QUAL文件为FASTQ文件¶
FASTQ 同时 包含序列和他们的质量信息字符串。FASTA文件 只 包含序列,而QUAL文件 只 包含质量。因此,一个单独的FASTQ文件可以转换为 成对的 FASTA和QUAL文件,FASTQ文件也可以由成对的FASTA和QUAL文件生成。
从FASTQ到FASTA很简单:
from Bio import SeqIO
SeqIO.convert("example.fastq", "fastq", "example.fasta", "fasta")
从FASTQ到QUAL也很简单:
from Bio import SeqIO
SeqIO.convert("example.fastq", "fastq", "example.qual", "qual")
然而,反向则有一点复杂。你可以使用 Bio.SeqIO.parse() 迭代一个 单独 文件中的所有记录,但是这里我们有两个输入文件。有几个可能的策略,然而这里假设两个文件是真的完全匹配的,最内存高效的方式是同时循环两个文件。代码有些巧妙,所以在 Bio.SeqIO.QualityIO 模块中我们提供一个函数来实现,叫做 PairedFastaQualIterator。它接受两个句柄(FASTA文件和QUAL文件)并返回一个 SeqRecord 迭代器:
from Bio.SeqIO.QualityIO import PairedFastaQualIterator
for record in PairedFastaQualIterator(open("example.fasta"), open("example.qual")):
print record
这个函数将检查FASTA和QUAL文件是否一致(例如,记录顺序是相同的,并且序列长度一致)。你可以和 Bio.SeqIO.write() 函数结合使用,转换一对FASTA和QUAL文件为单独的FASTQ文件:
from Bio import SeqIO
from Bio.SeqIO.QualityIO import PairedFastaQualIterator
handle = open("temp.fastq", "w") #w=write
records = PairedFastaQualIterator(open("example.fasta"), open("example.qual"))
count = SeqIO.write(records, handle, "fastq")
handle.close()
print "Converted %i records" % count
18.1.11 索引FASTQ文件¶
FASTQ文件通常非常大,包含上百万的读长。由于数据量的原因,你不能一次将所有的记录加载到内存中。这就是为什么上面的例子(过滤和剪切)以迭代的方式遍历整个文件,每次只查看一个 SeqRecord 。
然而,有时候你不能使用一个大的循环或迭代器 —— 你或许需要随机获取读长。这里 Bio.SeqIO.index() 函数被证明非常有用,它允许你使用名字获取FASTQ中的任何读长(参见章节 5.4.2 )。
我们将再次使用来自 ENA (ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz) 的文件 SRR020192.fastq ,尽管这是一个非常小的FASTQ文件,只有不到50,000读长:
>>> from Bio import SeqIO
>>> fq_dict = SeqIO.index("SRR020192.fastq", "fastq")
>>> len(fq_dict)
41892
>>> fq_dict.keys()[:4]
['SRR020192.38240', 'SRR020192.23181', 'SRR020192.40568', 'SRR020192.23186']
>>> fq_dict["SRR020192.23186"].seq
Seq('GTCCCAGTATTCGGATTTGTCTGCCAAAACAATGAAATTGACACAGTTTACAAC...CCG', SingleLetterAlphabet())
当在包含7百万读长的FASTQ文件上测试时,索引大概需要花费1分钟,然而获取记录几乎是瞬间完成的。
章节 18.1.5 的例子展示了如何使用 Bio.SeqIO.index() 函数来对FASTA文件进行排序 —— 这也可以用在FASTQ文件上。
18.1.12 转换SFF文件¶
如果你处理454(Roche)序列数据,你可能会接触Standard Flowgram Format (SFF)原始数据。这包括序列读长(called bases)、质量分数和原始流信息。
一个最常见的工作是转换SFF文件为一对FASTA和QUAL文件,或者一个单独的FASTQ文件。这可以使用 Bio.SeqIO.convert() 来轻松实现(参见 5.5.2 ):
>>> from Bio import SeqIO
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff", "reads.fasta", "fasta")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff", "reads.qual", "qual")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff", "reads.fastq", "fastq")
10
注意这个转换函数返回记录的条数,在这个例子中为10。这将给你 未裁剪 的读长,其中先导和跟随链中低质量的序列,或接头序列将以小写字母显示。如果你希望得到 裁剪 后的读长(使用SFF文件中的剪切信息),可以使用下面的代码:
>>> from Bio import SeqIO
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff-trim", "trimmed.fasta", "fasta")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff-trim", "trimmed.qual", "qual")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff-trim", "trimmed.fastq", "fastq")
10
如果你使用Linux,你可以向Roche请求一份“脱离仪器(off instrument)”的工具(通常叫做Newbler工具)。它提供了另一种的方式来在命令行实现SFF到FASTA或QUAL的转换(但并不支持FASTQ输出)。
$ sffinfo -seq -notrim E3MFGYR02_random_10_reads.sff > reads.fasta
$ sffinfo -qual -notrim E3MFGYR02_random_10_reads.sff > reads.qual
$ sffinfo -seq -trim E3MFGYR02_random_10_reads.sff > trimmed.fasta
$ sffinfo -qual -trim E3MFGYR02_random_10_reads.sff > trimmed.qual
Biopython以大小写混合的方式来表示剪切位点,这是有意模拟Roche工具的做法。
要获得Biopython对SFF支持的更多信息,请参考内部帮助:
>>> from Bio.SeqIO import SffIO
>>> help(SffIO)
...
18.1.13 识别开放读码框¶
在识别可能的基因中一个非常简单的第一步是寻找开放读码框(Open Reading Frame,ORF)。这里我们的意思是寻找六个编码框中所有的没有终止密码子的长区域 —— 一个ORF是一个不包含任何框内终止密码子的核苷酸区域。
当然,为了发现基因,你也需要确定起始密码子、可能的启动子的位置 —— 而且在真核生物中,你也需要关心内含子。然而,这种方法在病毒和原核生物中仍然有效。
为了展示怎样用Biopython实现这个目的,我们首先需要一个序列来查找。作为例子,我们再次使用细菌的质粒 —— 尽管这次我们将以没有任何基因标记的纯文本FASTA文件开始: NC_005816.fna 。这是一个细菌序列,所以我们需要使用NCBI密码子表11(参见章节 3.9 关于翻译的介绍)。
>>> from Bio import SeqIO
>>> record = SeqIO.read("NC_005816.fna","fasta")
>>> table = 11
>>> min_pro_len = 100
这里有一个巧妙的技巧,使用 Seq 对象的 split 方法获得一个包含六个读码框中所有可能的ORF翻译的列表:
>>> for strand, nuc in [(+1, record.seq), (-1, record.seq.reverse_complement())]:
... for frame in range(3):
... length = 3 * ((len(record)-frame) // 3) #Multiple of three
... for pro in nuc[frame:frame+length].translate(table).split("*"):
... if len(pro) >= min_pro_len:
... print "%s...%s - length %i, strand %i, frame %i" \
... % (pro[:30], pro[-3:], len(pro), strand, frame)
GCLMKKSSIVATIITILSGSANAASSQLIP...YRF - length 315, strand 1, frame 0
KSGELRQTPPASSTLHLRLILQRSGVMMEL...NPE - length 285, strand 1, frame 1
GLNCSFFSICNWKFIDYINRLFQIIYLCKN...YYH - length 176, strand 1, frame 1
VKKILYIKALFLCTVIKLRRFIFSVNNMKF...DLP - length 165, strand 1, frame 1
NQIQGVICSPDSGEFMVTFETVMEIKILHK...GVA - length 355, strand 1, frame 2
RRKEHVSKKRRPQKRPRRRRFFHRLRPPDE...PTR - length 128, strand 1, frame 2
TGKQNSCQMSAIWQLRQNTATKTRQNRARI...AIK - length 100, strand 1, frame 2
QGSGYAFPHASILSGIAMSHFYFLVLHAVK...CSD - length 114, strand -1, frame 0
IYSTSEHTGEQVMRTLDEVIASRSPESQTR...FHV - length 111, strand -1, frame 0
WGKLQVIGLSMWMVLFSQRFDDWLNEQEDA...ESK - length 125, strand -1, frame 1
RGIFMSDTMVVNGSGGVPAFLFSGSTLSSY...LLK - length 361, strand -1, frame 1
WDVKTVTGVLHHPFHLTFSLCPEGATQSGR...VKR - length 111, strand -1, frame 1
LSHTVTDFTDQMAQVGLCQCVNVFLDEVTG...KAA - length 107, strand -1, frame 2
RALTGLSAPGIRSQTSCDRLRELRYVPVSL...PLQ - length 119, strand -1, frame 2
注意,这里我们从 每条 序列的5’末(起始)端开始计算读码框。对 正向 链一直从5’末(起始)端开始计算有时更加容易。
你可以轻易编辑上面的循环代码,来创建一个待选蛋白列表,或者将它转换为一个列表解析。现在,这个代码所不能做的一个事情是记录蛋白的位置信息。
你可以用几种方式来处理。例如,下面的代码以蛋白计数的方式记录位置信息,并通过乘以三倍来转换为父序列(parent sequence),并记录编码框和链的信息:
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb","genbank")
table = 11
min_pro_len = 100
def find_orfs_with_trans(seq, trans_table, min_protein_length):
answer = []
seq_len = len(seq)
for strand, nuc in [(+1, seq), (-1, seq.reverse_complement())]:
for frame in range(3):
trans = str(nuc[frame:].translate(trans_table))
trans_len = len(trans)
aa_start = 0
aa_end = 0
while aa_start < trans_len:
aa_end = trans.find("*", aa_start)
if aa_end == -1:
aa_end = trans_len
if aa_end-aa_start >= min_protein_length:
if strand == 1:
start = frame+aa_start*3
end = min(seq_len,frame+aa_end*3+3)
else:
start = seq_len-frame-aa_end*3-3
end = seq_len-frame-aa_start*3
answer.append((start, end, strand,
trans[aa_start:aa_end]))
aa_start = aa_end+1
answer.sort()
return answer
orf_list = find_orfs_with_trans(record.seq, table, min_pro_len)
for start, end, strand, pro in orf_list:
print "%s...%s - length %i, strand %i, %i:%i" \
% (pro[:30], pro[-3:], len(pro), strand, start, end)
输出是:
NQIQGVICSPDSGEFMVTFETVMEIKILHK...GVA - length 355, strand 1, 41:1109
WDVKTVTGVLHHPFHLTFSLCPEGATQSGR...VKR - length 111, strand -1, 491:827
KSGELRQTPPASSTLHLRLILQRSGVMMEL...NPE - length 285, strand 1, 1030:1888
RALTGLSAPGIRSQTSCDRLRELRYVPVSL...PLQ - length 119, strand -1, 2830:3190
RRKEHVSKKRRPQKRPRRRRFFHRLRPPDE...PTR - length 128, strand 1, 3470:3857
GLNCSFFSICNWKFIDYINRLFQIIYLCKN...YYH - length 176, strand 1, 4249:4780
RGIFMSDTMVVNGSGGVPAFLFSGSTLSSY...LLK - length 361, strand -1, 4814:5900
VKKILYIKALFLCTVIKLRRFIFSVNNMKF...DLP - length 165, strand 1, 5923:6421
LSHTVTDFTDQMAQVGLCQCVNVFLDEVTG...KAA - length 107, strand -1, 5974:6298
GCLMKKSSIVATIITILSGSANAASSQLIP...YRF - length 315, strand 1, 6654:7602
IYSTSEHTGEQVMRTLDEVIASRSPESQTR...FHV - length 111, strand -1, 7788:8124
WGKLQVIGLSMWMVLFSQRFDDWLNEQEDA...ESK - length 125, strand -1, 8087:8465
TGKQNSCQMSAIWQLRQNTATKTRQNRARI...AIK - length 100, strand 1, 8741:9044
QGSGYAFPHASILSGIAMSHFYFLVLHAVK...CSD - length 114, strand -1, 9264:9609
如果你注释掉排序语句,那么蛋白序列将和之前显示的顺序一样,所以你能确定这是在做相同的事情。这里,我们可以按位置进行排序,使得和GenBank文件中的实际注释相比对较更加容易(就像章节 17.1.9 中显示的那样)。
然而,如果你想要的只是所有开放读码框的位置,翻译每一个可能的密码子将是很浪费时间的,包括转换和查找反向互补链。那么,你所要做的所有事情是查找可能的终止密码子(和他们反向互补)。使用正则表达式是一个很直接的方式(参见Python中的 re 模块)。这是描述查找字符串的一个非常强大的模块(然而非常复杂),也被许多编程语言和命令行工具,如 grep,所支持。你能找到一本书来描述它的使用!
18.2 序列解析与简单作图¶
这一部分展示更多使用第 5 章介绍的 Bio.SeqIO 模块进行序列解析的例子,以及Python类库matplotlib中 pylab 的作图接口(参见 matplotlib 主页的教程 )。注意,跟随这些例子,你需要安装matplotlib - 但是即使没有它,你依然可以尝试数据的解析的内容。
18.2.1 序列长度柱状图¶
在很多时候,你可能想要将某个数据集中的序列长度分布进行可视化 —— 例如,基因组组装项目中的contig的大小范围。在这个例子中,我们将再次使用我们的兰花FASTA文件 ls_orchid.fasta ,它只包含94条序列。
首先,我们使用 Bio.SeqIO 来解析这个FASTA文件,并创建一个序列长度的列表。你可以用一个for循环来实现,然而我觉得列表解析(list comprehension)更简洁:
>>> from Bio import SeqIO
>>> sizes = [len(rec) for rec in SeqIO.parse("ls_orchid.fasta", "fasta")]
>>> len(sizes), min(sizes), max(sizes)
(94, 572, 789)
>>> sizes
[740, 753, 748, 744, 733, 718, 730, 704, 740, 709, 700, 726, ..., 592]
现在我们得到了所有基因的长度(以整数列表的形式),我们可以用matplotlib的柱状图功能来显示它。
from Bio import SeqIO
sizes = [len(rec) for rec in SeqIO.parse("ls_orchid.fasta", "fasta")]
import pylab
pylab.hist(sizes, bins=20)
pylab.title("%i orchid sequences\nLengths %i to %i" \
% (len(sizes),min(sizes),max(sizes)))
pylab.xlabel("Sequence length (bp)")
pylab.ylabel("Count")
pylab.show()
这将弹出一个包含如下图形的新的窗口:
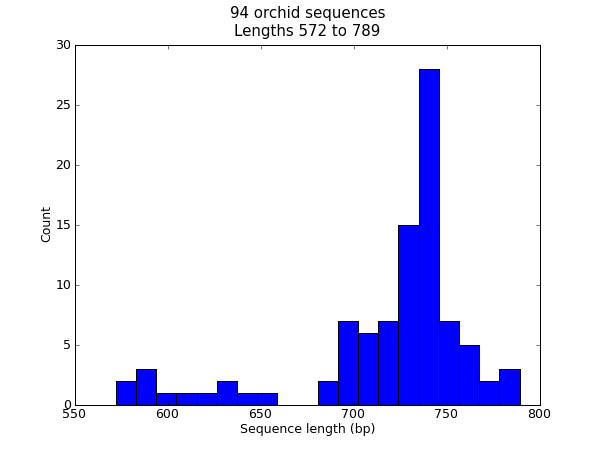
注意,这些兰花序列的长度大多数大约在740bp左右,这里有可能有两个不同长度的序列分类,其中包含一个较短的序列子集。
提示: 除了使用 pylab.show() 在窗口中显示图像以外,你也可以使用 pylab.savefig(...) 来将图像保存为图像文件中(例如PNG或PDF文件)。
18.2.2 序列GC%含量作图¶
对于核酸序列,另一个经常计算的值是GC含量。例如,你可能想要查看一个细菌基因组中所有基因的GC%,并研究任何离群值来确定可能最近通过基因水平转移而获得的基因。同样,对于这个例子,我们再次使用兰花FASTA文件 ls_orchid.fasta 。
首先,我们使用 Bio.SeqIO 解析这个FASTA文件并创建一个GC百分含量的列表。你可以使用for循环,但我更喜欢这样:
from Bio import SeqIO
from Bio.SeqUtils import GC
gc_values = sorted(GC(rec.seq) for rec in SeqIO.parse("ls_orchid.fasta", "fasta"))
读取完每个序列并计算了GC百分比,我们接着将它们按升序排列。现在,我们用matplotlib来对这个浮点数列表进行可视化:
import pylab
pylab.plot(gc_values)
pylab.title("%i orchid sequences\nGC%% %0.1f to %0.1f" \
% (len(gc_values),min(gc_values),max(gc_values)))
pylab.xlabel("Genes")
pylab.ylabel("GC%")
pylab.show()
像之前的例子一样,弹出一个窗口中将包含如下图形:
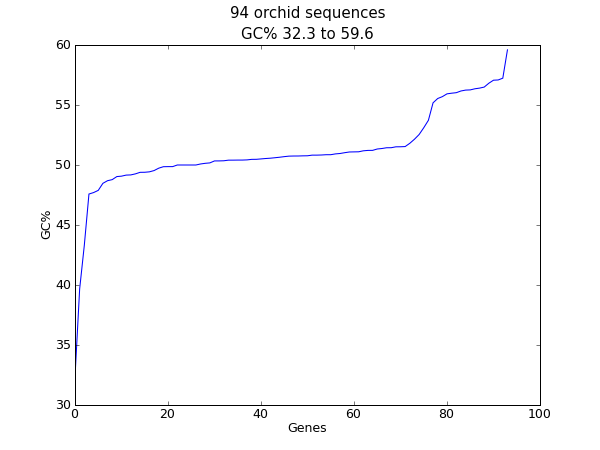
如果你使用的是一个物种中的所有基因集,你可能会得到一个更加平滑的图。
18.2.3 核苷酸点线图¶
点线图是可视化比较两条核苷酸序列的相似性的一种方式。采用一个滑动窗来相互比较较短的子序列(比较通常根据一个不匹配阈值来实现)。为了简单起见,此处我们将只查找完全匹配(如下图黑色所示)。
我们需要两条序列开始。为了论证,我们只取兰花FASTA文件中的前两条序列。ls_orchid.fasta:
from Bio import SeqIO
handle = open("ls_orchid.fasta")
record_iterator = SeqIO.parse(handle, "fasta")
rec_one = record_iterator.next()
rec_two = record_iterator.next()
handle.close()
我们将展示两种方式。首先,一个简单的实现,它将所有滑动窗大小的子序列相互比较,并生产一个相似性矩阵。你可以创建一个矩阵或数组对象,而在这儿,我们只用一个用嵌套的列表解析生成的布尔值列表的列表。
window = 7
seq_one = str(rec_one.seq).upper()
seq_two = str(rec_two.seq).upper()
data = [[(seq_one[i:i+window] <> seq_two[j:j+window]) \
for j in range(len(seq_one)-window)] \
for i in range(len(seq_two)-window)]
注意,我们在这里并 没有 检查反向的互补匹配。现在我们将使用matplotlib的 pylab.imshow() 函数来显示这个数据,首先启用灰度模式,以保证这是在黑白颜色下完成的:
import pylab
pylab.gray()
pylab.imshow(data)
pylab.xlabel("%s (length %i bp)" % (rec_one.id, len(rec_one)))
pylab.ylabel("%s (length %i bp)" % (rec_two.id, len(rec_two)))
pylab.title("Dot plot using window size %i\n(allowing no mis-matches)" % window)
pylab.show()
这将弹出一个新的窗口,包含类似这样的图形:
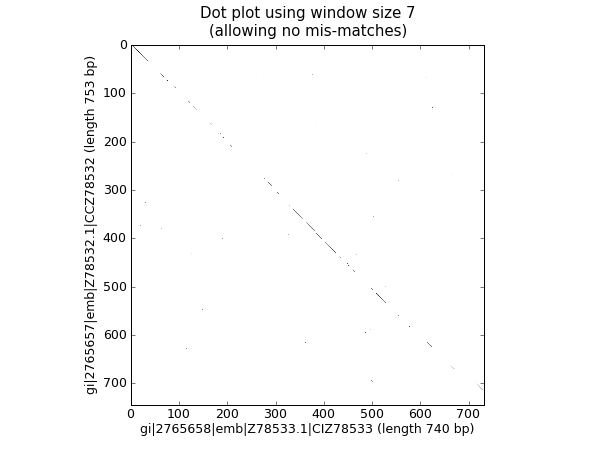
可能如您所料,这两条序列非常相似,图中部分滑动窗大小的线沿着对角线匹配。图中没有对角线外的点,这意味着序列中并没有倒位或其他有趣的偏离对角线匹配。
上面的代码在小的例子中工作得很好,但是应用到大的序列时,这里有两个问题。首先,这种以穷举地方式进行所有可能的两两比对非常缓慢。作为替代,我们将创建一个词典来映射所有滑动窗大小的子序列的位置,然后取两者的交集来获得两条序列中都发现的子序列。这将占用更多的内存,然而速度 更 快。另外, pylab.imshow() 函数只能显示较小的矩阵。作为替代,我们将使用 pylab.scatter() 函数。
我们从创建,从滑动窗大小的子序列到其位置的字典映射开始:
window = 7
dict_one = {}
dict_two = {}
for (seq, section_dict) in [(str(rec_one.seq).upper(), dict_one),
(str(rec_two.seq).upper(), dict_two)]:
for i in range(len(seq)-window):
section = seq[i:i+window]
try:
section_dict[section].append(i)
except KeyError:
section_dict[section] = [i]
#Now find any sub-sequences found in both sequences
#(Python 2.3 would require slightly different code here)
matches = set(dict_one).intersection(dict_two)
print "%i unique matches" % len(matches)
为了使用 pylab.scatter() 函数,我们需要两个分别对应 x 和 y 轴的列表:
#Create lists of x and y co-ordinates for scatter plot
x = []
y = []
for section in matches:
for i in dict_one[section]:
for j in dict_two[section]:
x.append(i)
y.append(j)
现在我们能以散点图的形式画出优化后的点线图:
import pylab
pylab.cla() #clear any prior graph
pylab.gray()
pylab.scatter(x,y)
pylab.xlim(0, len(rec_one)-window)
pylab.ylim(0, len(rec_two)-window)
pylab.xlabel("%s (length %i bp)" % (rec_one.id, len(rec_one)))
pylab.ylabel("%s (length %i bp)" % (rec_two.id, len(rec_two)))
pylab.title("Dot plot using window size %i\n(allowing no mis-matches)" % window)
pylab.show()
这将弹出一个新的窗口,包含如下图形:
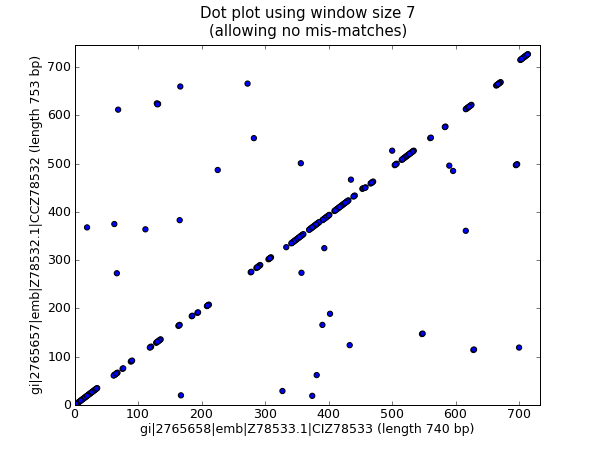
我个人认为第二个图更加易读!再次注意,我们在这里 没有 检查反向互补匹配 —— 你可以扩展这个例子来实现它,或许可以以一种颜色显示正向匹配,另一种显示反向匹配。
18.2.4 绘制序列读长数据的质量图¶
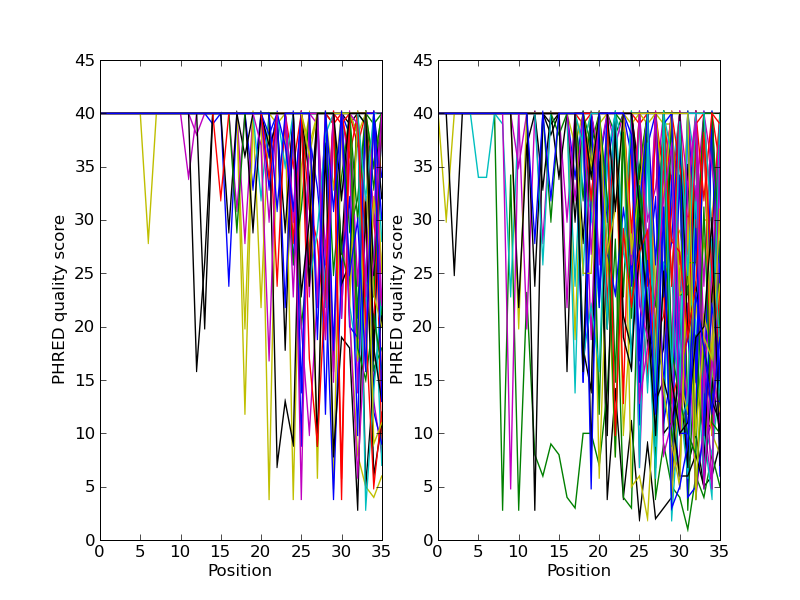
如果你在处理二代测序数据,你可能希望绘制数据的质量图。这里使用两个包含双末端(paired end)读长的FASTQ文件作为例子,其中 SRR001666_1.fastq 为正向读长, SRR001666_2.fastq 为反向读长。它们可以从ENA序列读长档案的FTP站点下载( ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR001/SRR001666/SRR001666_1.fastq.gz 和 ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR001/SRR001666/SRR001666_2.fastq.gz ), 且来自 E. coli —— 参见 http://www.ebi.ac.uk/ena/data/view/SRR001666 的详细介绍。在下面的代码中, pylab.subplot(...) 函数被用来在两个子图中展示正向和反向的质量。这里也有少量的代码来保证仅仅展示前50个读长的质量。
import pylab
from Bio import SeqIO
for subfigure in [1,2]:
filename = "SRR001666_%i.fastq" % subfigure
pylab.subplot(1, 2, subfigure)
for i,record in enumerate(SeqIO.parse(filename, "fastq")):
if i >= 50 : break #trick!
pylab.plot(record.letter_annotations["phred_quality"])
pylab.ylim(0,45)
pylab.ylabel("PHRED quality score")
pylab.xlabel("Position")
pylab.savefig("SRR001666.png")
print "Done"
你应该注意到,这里我们使用了 Bio.SeqIO 的格式名称 fastq ,因为NCBI使用标准Sanger FASTQ和PHRED分数的存储这些读长。然而,你可能从读长的长度中猜到,这些数据来自Illumina Genome Analyzer,而且可能最初是以Solexa/Illumina FASTQ两种格式变种中的一种存在。
这个例子使用 pylab.savefig(...) 函数,而不是``pylab.show(...)`` ,然而就像前面提到的一样,它们两者都非常有用。下面是得到的结果:
18.3 处理序列比对¶
这部分可以看做是第 6 章的继续。
18.3.1 计算摘要信息¶
一旦你有一个比对,你很可能希望找出关于它的一些信息。我们尽力将这些功能分离到单独的能作用于比对对象的类中,而不是将所有的能生成比对信息的函数都放入比对对象本身。
准备计算比对对象的摘要信息非常快捷。假设我们已经得到了一个比对对象 alignment ,例如由在第 6 章介绍的 Bio.AlignIO.read(...) 读入。我们获得该对象的摘要信息所要做的所有事情是:
from Bio.Align import AlignInfo
summary_align = AlignInfo.SummaryInfo(alignment)
summary_align 对象非常有用,它将帮你做以下巧妙的事情:
18.3.2 计算一个快速一致序列¶
在章节 18.3.1 中描述的 SummaryInfo 对象提供了一个可以快速计算比对的保守(consensus)序列的功能。假设我们有一个 SummaryInfo 对象,叫做 summary_align,我们能通过下面的方法计算一个保守序列:
consensus = summary_align.dumb_consensus()
就行名字显示的那样,这是一个非常简单的保守序列计算器,它将只是在保守序列中累加每个位点的所有残基,如果最普遍的残基数大于某个阈值时,这个最普遍的残基将被添加到保守序列中。如果它没有到达这个阈值,将添加一个“不确定字符”。最终返回的保守序列对象是一个Seq对象,它的字母表是从组成保守序列所有序列的字母表中推断出来的。所以使用 print consensus 将给出如下信息:
consensus Seq('TATACATNAAAGNAGGGGGATGCGGATAAATGGAAAGGCGAAAGAAAGAAAAAAATGAAT
...', IUPACAmbiguousDNA())
你可以通过传入可选参数来调整 dumb_consensus 的工作方式:
- the threshold
- 这是用来设定某个残基在某个位点出现的频率超过一定阈值,才将其添加到保守序列。默认为0.7(即70%)。
- the ambiguous character
- 指定保守序列中的不确定字符。默认为’N’。
- the consensus alphabet
- 指定保守序列的字母表。如果没有提供,我们将从比对序列的字母表基础上推断该字母表。
18.3.3 位点特异性评分矩阵¶
位点特异性评分矩阵(Position specific score matrices,PSSMs)以另一种总结比对信息的方式(与刚才介绍的保守序列不同),这或许在某些情况下更为有用。简单来说,PSSM是一个计数矩阵。对于比对中的每一列,将所有可能出现的字母进行计数并加和。这些加和值将和一个代表序列(默认为比对中的第一条序列)一起显示出来。这个序列可能是保守序列,但也可以是比对中的任何序列。例如,对于比对,
GTATC
AT--C
CTGTC
它的PSSM是:
G A T C
G 1 1 0 1
T 0 0 3 0
A 1 1 0 0
T 0 0 2 0
C 0 0 0 3
假设我们有一个比对对象叫做 c_align ,为了获得PSSM和保守序列,我们首先得到一个摘要对象(summary object),并计算一致序列:
summary_align = AlignInfo.SummaryInfo(c_align)
consensus = summary_align.dumb_consensus()
现在,我们想创建PSSM,但是在计算中忽略任何 N 不确定残基:
my_pssm = summary_align.pos_specific_score_matrix(consensus,
chars_to_ignore = ['N'])
关于此有亮点需要说明:
为了维持字母表的严格性,你可以在PSSM的顶部显示比对对象字母表中规定的字符。空白字符(Gaps)并不包含在PSSM的顶轴中。
传入并显示在左侧轴的序列可以不是保守序列。例如,你如果想要在PSSM左边显示比对中的第二条序列,你只需要:
second_seq = alignment.get_seq_by_num(1) my_pssm = summary_align.pos_specific_score_matrix(second_seq chars_to_ignore = ['N'])
以上的命令将返回一个 PSSM 对象。为了显示出PSSM,我们只需 print my_pssm,结果如下:
A C G T
T 0.0 0.0 0.0 7.0
A 7.0 0.0 0.0 0.0
T 0.0 0.0 0.0 7.0
A 7.0 0.0 0.0 0.0
C 0.0 7.0 0.0 0.0
A 7.0 0.0 0.0 0.0
T 0.0 0.0 0.0 7.0
T 1.0 0.0 0.0 6.0
...
你可以用 your_pssm[sequence_number][residue_count_name] 获得任何PSSM的元素。例如,获取上面PSSM中第二个元素的‘A’残基的计数,你可以:
>>> print my_pssm[1]["A"]
7.0
PSSM类的结构有望使得获取元素和打印漂亮的矩阵都很方便。
18.3.4 信息量¶
一个潜在而有用的衡量进化保守性的测度是序列的信息量。
一个有用的针对分子生物学家的信息论的介绍可以在这里找到: http://www.lecb.ncifcrf.gov/~toms/paper/primer/ 。对于我们的目地,我们将查看保守序列或其部分序列的信息量。我们使用下面的公式计算多序列比对中某个特定的列的信息量:
其中:
- IC*j* – 比对中第 j 列的信息量。
- N*a* – 字母表中字母的个数。
- P*ij* – 第 j 列的某个特定字母 i 的频率(即,如果G在包含有6个序列的比对中有3次出现,则该列G的信息量为0.5)
- Q*i* – 字母 i 的期望频率。这是一个可选参数,由用户自行决定使用。默认情况下,它被自动赋值为0.05 = 1/20,若为蛋白字母表;或0.25 = 1/4 ,若为核酸字母表。这是在没有先验分布假设的情况下计算信息量。而在假设先验分布或使用非标准字母表时,你需要提供 Q*i* 的值。
好了,现在我们知道Biopython如何计算了序列比对的信息量,让我们看看怎么对部分比对区域进行计算。
首先,我们需要使用我们的比对来获得一个比对摘要对象,我们假设它叫做 summary_align (参见章节 18.3.1 来了解怎样得到它)。一旦我们得到这个对象,计算某个区域的信息量就像下面一样简单:
info_content = summary_align.information_content(5, 30,
chars_to_ignore = ['N'])
哇哦,这比上面的公式看起来要简单多了!变量 info_content 现在含有一个浮点数来表示指定区域(比对中的5到30)的信息量。我们在计算信息量时特意忽略了不确定残基’N’,因为这个值没有包括在我们的字母表中(因而我们不必要关心它!)。
像上面提到的一样,我们同样能通过提供期望频率计算相对信息量:
expect_freq = {
'A' : .3,
'G' : .2,
'T' : .3,
'C' : .2}
期望值不能以原始的字典传入,而需要作为 SubsMat.FreqTable 对象传入(参见章节 20.2.2 以获得关于FreqTables的更多信息)。FreqTable对象提供了一个关联字典和字母表的标准,这和Biopython中Seq类的工作方式类似。
要从频率字典创建一个FreqTable对象,你只需要:
from Bio.Alphabet import IUPAC
from Bio.SubsMat import FreqTable
e_freq_table = FreqTable.FreqTable(expect_freq, FreqTable.FREQ,
IUPAC.unambiguous_dna)
现在我们得到了它,计算我们比对区域的相对信息量就像下面一样简单:
info_content = summary_align.information_content(5, 30,
e_freq_table = e_freq_table,
chars_to_ignore = ['N'])
现在,info_content 将包含与期望频率相关的该区域的相对信息量。
返回值是按上面的公式以2为对底数计算的。你可以通过传入 log_base 参数来改变成你想要的底数:
info_content = summary_align.information_content(5, 30, log_base = 10,
chars_to_ignore = ['N'])
好了,现在你已经知道怎么计算信息量了。如果你想要在实际的生命科学问题中应用它,最好找一些关于 信息量的文献钻研,以了解它是怎样用的。希望你的钻研不会发现任何有关这个函数的编码错误。
18.4 替换矩阵¶
替换矩阵是每天的生物信息学工作中的极端重要的一部分。它们提供决定两个不同的残基有多少相互替换的可能性的得分规则。这在序列比较中必不可少。Durbin等的“Biological Biological Sequence Analysis” 一书中提供了对替换矩阵以及它们的用法的非常好的介绍。一些非常有名的替换矩阵是PAM和BLOSUM系列矩阵。
Biopython提供了大量的常见替换矩阵,也提供了创建你自己的替换矩阵的功能。
18.4.1 使用常见替换矩阵¶
18.4.2 从序列比对创建你自己的替换矩阵¶
使用替换矩阵类能轻易做出的一个非常酷的事情,是从序列比对创建出你自己的替换矩阵。实际中,通常是使用蛋白比对来做。在这个例子中,我们将首先得到一个Biopython比对对象,然后得到一个摘要对象来计算关于这个比对的相关信息。文件 protein.aln (也可在 这里 获取)包含Clustalw格式的比对输出。
>>> from Bio import AlignIO
>>> from Bio import Alphabet
>>> from Bio.Alphabet import IUPAC
>>> from Bio.Align import AlignInfo
>>> filename = "protein.aln"
>>> alpha = Alphabet.Gapped(IUPAC.protein)
>>> c_align = AlignIO.read(filename, "clustal", alphabet=alpha)
>>> summary_align = AlignInfo.SummaryInfo(c_align)
章节 6.4.1 和 18.3.1 包含关于此类做法的更多信息。
现在我们得到了我们的 summary_align 对象,我们想使用它来找出不同的残基相互替换的次数。为了使例子的可读性更强,我们将只关注那些有极性电荷侧链的氨基酸。幸运的是,这能在生成替代字典时轻松实现,通过传入所有需要被忽略的字符。这样我们将能创建一个只包含带电荷的极性氨基酸的替代字典:
>>> replace_info = summary_align.replacement_dictionary(["G", "A", "V", "L", "I",
... "M", "P", "F", "W", "S",
... "T", "N", "Q", "Y", "C"])
这个关于氨基酸替代的信息以python字典的形式展示出来将会像如下的样子(顺序可能有所差异):
{('R', 'R'): 2079.0, ('R', 'H'): 17.0, ('R', 'K'): 103.0, ('R', 'E'): 2.0,
('R', 'D'): 2.0, ('H', 'R'): 0, ('D', 'H'): 15.0, ('K', 'K'): 3218.0,
('K', 'H'): 24.0, ('H', 'K'): 8.0, ('E', 'H'): 15.0, ('H', 'H'): 1235.0,
('H', 'E'): 18.0, ('H', 'D'): 0, ('K', 'D'): 0, ('K', 'E'): 9.0,
('D', 'R'): 48.0, ('E', 'R'): 2.0, ('D', 'K'): 1.0, ('E', 'K'): 45.0,
('K', 'R'): 130.0, ('E', 'D'): 241.0, ('E', 'E'): 3305.0,
('D', 'E'): 270.0, ('D', 'D'): 2360.0}
这个信息提供了我们所需要的替换次数,或者说我们期望的不同的事情相互替换有多么频繁。事实上,(你可能会感到惊奇)这就是我们继续创建替代矩阵所需要的全部信息。首先,我们使用替代字典信息创建一个“接受替换矩阵”(Accepted Replacement Matrix,ARM):
>>> from Bio import SubsMat
>>> my_arm = SubsMat.SeqMat(replace_info)
使用这个“接受替换矩阵”,我们能继续创建我们的对数矩阵(即一个标准类型的替换举证):
>>> my_lom = SubsMat.make_log_odds_matrix(my_arm)
在创建的这个对数矩阵时有以下可选参数:
exp_freq_table– 你可以传入一个每个字母的期望频率的表格。如果提供,在计算期望替换时,这将替代传入的“接收替换矩阵”。logbase- 用来创建对数奇数矩阵的对数底数。默认为10。factor- 用来乘以每个矩阵元素的因数。默认为10,这样通常可以使得矩阵的数据容易处理。round_digit- 矩阵中四舍五入所取的小数位数,默认为0(即没有小数)。
一旦你获得了你的对数矩阵,你可以使用函数 print_mat 很漂亮的显示出来。使用我们创建的矩阵可以得到:
>>> my_lom.print_mat()
D 2
E -1 1
H -5 -4 3
K -10 -5 -4 1
R -4 -8 -4 -2 2
D E H K R
很好。我们现在得到了自己的替换矩阵!
18.5 BioSQL – 存储序列到关系数据库中¶
BioSQL 是 OBF 多个项目(BioPerl、 BioJava等)为了支持共享的存储序列数据的数据库架构而共同努力的结果。理论上,你可以用BioPerl加载GenBank文件到数据库中,然后用Biopython从数据库中提取出来为一个包含Feature的Record对象 —— 并获得或多或少和直接用 Bio.SeqIO (第 5 章)加载GenBank文件为SeqRecord相同的东西。
Biopython中BioSQL模块的文档目前放在 http://biopython.org/wiki/BioSQL ,是我们维基页面的一部分。