第11章 走向3D:PDB模块¶
Bio.PDB是Biopython中处理生物大分子晶体结构的模块。除了别的类之外,Bio.PDB包含PDBParser类,此类能够产生一个Structure对象,以一种较方便的方式获取文件中的原子数据。只是在处理PDB文件头所包含的信息时,该类有一定的局限性。
11.1 晶体结构文件的读与写¶
11.1.1 读取PDB文件¶
首先,我们创建一个 PDBParser 对象:
>>> from Bio.PDB.PDBParser import PDBParser
>>> p = PDBParser(PERMISSIVE=1)
PERMISSIV 标签表示一些与PDB文件相关的问题(见 11.7.1 )会被忽略(注意某些原子和/或残基会丢失)。如果没有这个标签,则会在解析器运行期间有问题被检测到的时候生成一个 PDBConstructionException 标签。
接着通过 PDBParser 解析PDB文件,就产生了Structure对象(在此例子中,PDB文件为’pdb1fat.ent’,‘1fat’是用户定义的结构名称):
>>> structure_id = "1fat"
>>> filename = "pdb1fat.ent"
>>> s = p.get_structure(structure_id, filename)
你可以从PDBParser对象中用 get_header 和 get_trailer 方法来提取PDB文件中的文件头和文件尾(简单的字符串列表)。然而许多PDB文件头包含不完整或错误的信息。许多错误在等价的mmCIF格式文件中得到修正。* 因此,如果你对文件头信息感兴趣,可以用下面即将讲到的 MMCIF2Dict 来提取信息,而不用处理PDB文件文件头。*
现在澄清了,让我们回到解析PDB文件头这件事上。结构对象有个属性叫 header ,这是一个将头记录映射到其相应值的Python字典。
例子:
>>> resolution = structure.header['resolution']
>>> keywords = structure.header['keywords']
在这个字典中可用的关键字有 name 、 head 、 deposition_date 、 release_date 、 structure_method 、 resolution 、 structure_reference (映射到一个参考文献列表)、 journal_reference 、 author 、和 compound (映射到一个字典,其中包含结晶化合物的各种信息)。
没有创建 Structure 对象的时候,也可以创建这个字典,比如直接从PDB文件创建:
>>> file = open(filename,'r')
>>> header_dict = parse_pdb_header(file)
>>> file.close()
11.1.2 读取mmCIF文件¶
与PDB文件的情形类似,先创建一个 MMCIFParser 对象:
>>> from Bio.PDB.MMCIFParser import MMCIFParser
>>> parser = MMCIFParser()
然后用这个解析器从mmCIF文件创建一个结构对象:
>>> structure = parser.get_structure('1fat', '1fat.cif')
为了尽量少访问mmCIF文件,可以用 MMCIF2Dict 类创建一个Python字典来将所有mmCIF文件中各种标签映射到其对应的值上。若有多个值(像 _atom_site.Cartn_y 标签,储存的是所有原子的*y*坐标值),则这个标签映射到一个值列表。从mmCIF文件创建字典如下:
>>> from Bio.PDB.MMCIF2Dict import MMCIF2Dict
>>> mmcif_dict = MMCIF2Dict('1FAT.cif')
例:从mmCIF文件获取溶剂含量:
>>> sc = mmcif_dict['_exptl_crystal.density_percent_sol']
例:获取包含所有原子*y*坐标的列表:
>>> y_list = mmcif_dict['_atom_site.Cartn_y']
11.1.3 读取PDB XML格式的文件¶
这个功能暂时还不支持,不过我们确实计划在未来支持这个功能(这项任务并不大)。如果你需要的话联系Biopython开发人员( biopython-dev@biopython.org )。
11.1.4 写PDB文件¶
可以用PDBIO类实现。当然也可很方便地输出一个结构的特定部分。
例子:保存一个结构
>>> io = PDBIO()
>>> io.set_structure(s)
>>> io.save('out.pdb')
如果你想写出结构的一部分,可以用 Select 类(也在 PDBIO 中)来实现。 Select 有如下四种方法:
accept_model(model)accept_chain(chain)accept_residue(residue)accept_atom(atom)
在默认情况下,每种方法的返回值都为1(表示model/chain/residue/atom被包含在输出结果中)。通过子类化 Select 和返回值0,你可以从输出中排除model、chain等。也许麻烦,但很强大。接下来的代码将只输出甘氨酸残基:
>>> class GlySelect(Select):
... def accept_residue(self, residue):
... if residue.get_name()=='GLY':
... return True
... else:
... return False
...
>>> io = PDBIO()
>>> io.set_structure(s)
>>> io.save('gly_only.pdb', GlySelect())
如果这部分对你来说太复杂,那么 Dice 模块有一个很方便的 extract 函数,它可以输出一条链中起始和终止氨基酸残基之间的所有氨基酸残基。
11.2 结构的表示¶
一个 Structure 对象的整体布局遵循称为SMCRA(Structure/Model/Chain/Residue/Atom,结构/模型/链/残基/原子)的体系架构:
- 结构由模型组成
- 模型由多条链组成
- 链由残基组成
- 多个原子构成残基
这是很多结构生物学家/生物信息学家看待结构的方法,也是处理结构的一种简单而有效的方法。在需要的时候加上额外的材料。一个 Structure 对象的UML图(暂时忘掉 Disordered 吧)如下图所示 11.1 。这样的数据结构不一定最适用于表示一个结构的生物大分子内容,但要很好地解释一个描述结构的文件中所呈现的数据(最典型的如PDB或MMCIF文件),这样的数据结构就是必要的了。如果这种层次结构不能表示一个结构文件的内容,那么可以相当确定是这个文件有错误或至少描述结构不够明确。一旦不能生成SMCRA数据结构,就有理由怀疑出了故障。因此,解析PDB文件可用于检测可能的故障。我们将在 11.7.1 小节给出关于这一点的一些例子。
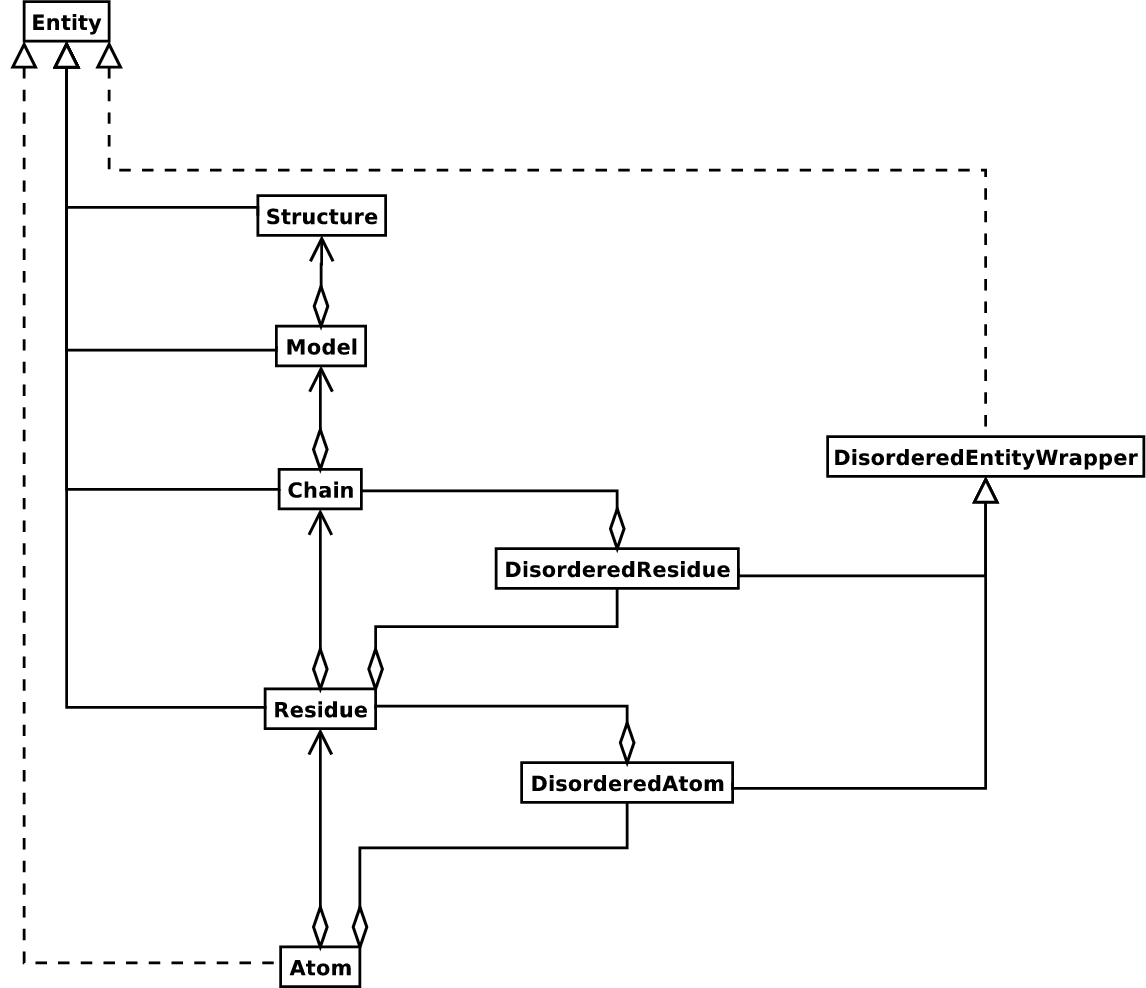
图11.1:用来表示大分子结构的 Structure 类的SMCRA体系的UML图。带方块的实线表示集合,带箭头的实线表示引用,带三角形的实线表示继承,带三角形的虚线表示接口实现。
结构,模型,链,残基都是实体基类的子类。原子类仅仅(部分)实现了实体接口(因为原子类没有子类)。
对于每个实体子类,你可以用该子类的一个唯一标识符作为键来提取子类(比如,可以用原子名称作为键从残基对象中提取一个原子对象;用链的标识符作为键从域对象中提取链)。
紊乱原子和残基用DisorderedAtom和DisorderedResidue类来表示,二者都是DisorderedEntityWrapper基类的子类。它们隐藏了紊乱的复杂性,表现得与原子和残基对象无二。
一般地,一个实体子类(即原子,残基,链,模型)能通过标识符作为键来从父类(分别为残基,链,模型,结构)中提取。
>>> child_entity = parent_entity[child_id]
你可以从一个父实体对象获得所有子实体的列表。需要注意的是,这个列表以一种特定的方式排列(例如根据在模型对象中链对象的链标识符来排序)。
>>> child_list = parent_entity.get_list()
你也可以从子类得到父类:
>>> parent_entity = child_entity.get_parent()
在SMCRA的所有层次水平,你还可以提取一个 完整id 。完整id是包含所有从顶层对象(结构)到当前对象的id的一个元组。一个残基对象的完整id可以这么得到:
>>> full_id = residue.get_full_id()
>>> print full_id
("1abc", 0, "A", ("", 10, "A"))
这对应于:
- id为”1abc”的结构
- id为0的模型
- id为”A”的链
- id为(” ”, 10, “A”)的残基
这个残基id表示该残基不是异质残基(也不是水分子),因为其异质值为空;而序列标识符为10,插入码为”A”。
要得到实体的id,用 get_id 方法即可:
>>> entity.get_id()
可以用 has_id 方法来检查这个实体是否有子类具有给定id:
>>> entity.has_id(entity_id)
实体的长度等于其子类的个数:
>>> nr_children = len(entity)
对于从父实体得到的子实体,可以删除,重命名,添加等等,但这并不包含任何完整性检查(比如,有可能添加两个相同id的残基到同一条链上)。这就真的需要包含完整性检查的装饰类(Decorator)来完成了,但是如果你想使用原始接口的话可以查看源代码(Entity.py)。
11.2.1 结构¶
结构对象是层次中的最高层。其id是用户指定的一个字符串。结构包含一系列子模型。大部分晶体结构(但不是全部)含有一个单一模型,但是NMR结构通常由若干模型构成。晶体结构中大部分子的乱序也能导致多个模型。
11.2.2 模型¶
结构域对象的id是一个整数,源自该模型在所解析文件中的位置(自动从0开始)。晶体结构通常只有一个模型(id为0),而NMR文件通常含有多个模型。然而许多PDB解析器都假定只有一个结构域, Bio.PDB 中的 Structure 类就设计成能轻松处理含有不止一个模型的PDB文件。
举个例子,从一个结构对象中获取其第一个模型:
>>> first_model = structure[0]
模型对象存储着子链的列表。
11.2.3 链¶
链对象的id来自PDB/mmCIF文件中的链标识符,是个单字符(通常是一个字母)。模型中的每个链都具有唯一的id。例如,从一个模型对象中取出标识符为“A”的链对象:
>>> chain_A = model["A"]
链对象储存着残基对象的列表。
11.2.4 残基¶
一个残基id是一个三元组:
异质域 (hetfield),即:
'W'代表水分子'H_'后面紧跟残基名称,代表其它异质残基(例如'H_GLC'表示一个葡萄糖分子)- 空值表示标准的氨基酸和核酸
采用这种体制的理由在 11.4.1 部分有叙述。
序列标识符 (resseq),一个描述该残基在链上的位置的整数(如100);
插入码 (icode),一个字符串,如“A”。插入码有时用来保存某种特定的、想要的残基编号体制。一个Ser 80的插入突变(比如在Thr 80和Asn 81残基间插入)可能具有如下序列标识符和插入码:Thr 80 A, Ser 80 B, Asn 81。这样一来,残基编号体制保持与野生型结构一致。
因此,上述的葡萄酸残基id就是 (’H_GLC’, 100, ’A’) 。如果异质标签和插入码为空,那么可以只使用序列标识符:
# Full id
>>> residue=chain[(' ', 100, ' ')]
# Shortcut id
>>> residue=chain[100]
异质标签的起因是许许多多的PDB文件使用相同的序列标识符表示一个氨基酸和一个异质残基或一个水分子,这会产生一个很明显的问题,如果不使用异质标签的话。
毫不奇怪,一个残基对象存储着一个子原子集,它还包含一个表示残基名称的字符串(如 “ASN”)和残基的片段标识符(这对X-PLOR的用户来说很熟悉,但是在SMCRA数据结构的构建中没用到)。
让我们来看一些例子。插入码为空的Asn 10具有残基id (’ ’, 10, ’ ’) ;Water 10,残基id (’W’, 10, ’ ’);一个序列标识符为10的葡萄糖分子(名称为GLC的异质残基),残基id为 (’H_GLC’, 10, ’ ’) 。在这种情况下,三个残基(具有相同插入码和序列标识符)可以位于同一条链上,因为它们的残基id是不同的。
大多数情况下,hetflag和插入码均为空,如 (’ ’, 10, ’ ’) 。在这些情况下,序列标识符可以用作完整id的快捷方式:
# use full id
>>> res10 = chain[(' ', 10, ' ')]
# use shortcut
>>> res10 = chain[10]
一个链对象中每个残基对象都应该具有唯一的id。但是对含紊乱原子的残基,要以一种特殊的方式来处理,详见 11.3.3 。
一个残基对象还有大量其它方法:
>>> residue.get_resname() # returns the residue name, e.g. "ASN"
>>> residue.is_disordered() # returns 1 if the residue has disordered atoms
>>> residue.get_segid() # returns the SEGID, e.g. "CHN1"
>>> residue.has_id(name) # test if a residue has a certain atom
你可以用 is_aa(residue) 来检验一个残基对象是否为氨基酸。
11.2.5 原子¶
原子对象储存着所有与原子有关的数据,它没有子类。原子的id就是它的名称(如,“OG”代表Ser残基的侧链氧原子)。在残基中原子id必需是唯一的。此外,对于紊乱原子会产生异常,见 11.3.2 小节的描述。
原子id就是原子名称(如 ’CA’ )。在实践中,原子名称是从PDB文件中原子名称去除所有空格而创建的。
但是在PDB文件中,空格可以是原子名称的一部分。通常,钙原子称为 ’CA..’ 是为了和Cα原子(叫做 ’.CA.’ )区分开。在这种情况下,如果去掉空格就会产生问题(如统一个残基中的两个原子都叫做 ’CA’ ),所以保留空格。
在PDB文件中,一个原子名字由4个字符组成,通常头尾皆为空格。为了方便使用,空格通常可以去掉(在PDB文件中氨基酸的Cα原子标记为“.CA.”,点表示空格)。为了生成原子名称(然后是原子id),空格删掉了,除非会在一个残基中造成名字冲突(如两个原子对象有相同的名称和id)。对于后面这种情况,会尝试让原子名称包含空格。这种情况可能会发生在,比如残基包含名称为“.CA.”和“CA..”的原子,尽管这不怎么可能。
所存储的原子数据包括原子名称,原子坐标(如果有的话还包括标准差),B因子(包括各向异性B因子和可能存在的标准差),altloc标识符和完整的、包括空格的原子名称。较少用到的项如原子序号和原子电荷(有时在PDB文件中规定)也就没有存储。
为了处理原子坐标,可以用 ’Atom’ 对象的 transform 方法。用 set_coord 方法可以直接设定原子坐标。
一个Atom对象还有如下其它方法:
>>> a.get_name() # atom name (spaces stripped, e.g. "CA")
>>> a.get_id() # id (equals atom name)
>>> a.get_coord() # atomic coordinates
>>> a.get_vector() # atomic coordinates as Vector object
>>> a.get_bfactor() # isotropic B factor
>>> a.get_occupancy() # occupancy
>>> a.get_altloc() # alternative location specifier
>>> a.get_sigatm() # standard deviation of atomic parameters
>>> a.get_siguij() # standard deviation of anisotropic B factor
>>> a.get_anisou() # anisotropic B factor
>>> a.get_fullname() # atom name (with spaces, e.g. ".CA.")
siguij,各向异性B因子和sigatm Numpy阵列可以用来表示原子坐标。
get_vector 方法会返回一个代表 Atom 对象坐标的 Vector 对象,可以对原子坐标进行向量运算。 Vector 实现了完整的三维向量运算、矩阵乘法(包括左乘和右乘)和一些高级的、与旋转相关的操作。
举个Bio.PDB的 Vector 模块功能的例子,假设你要查找Gly残基的Cβ原子的位置,如果存在的话。将Gly残基的N原子沿Cα-C化学键旋转-120度,能大致将其放在一个真正的Cβ原子的位置上。怎么做呢?就是下面这样使用 Vector 模块中的``rotaxis`` 方法(能用来构造一个绕特定坐标轴的旋转):
# get atom coordinates as vectors
>>> n = residue['N'].get_vector()
>>> c = residue['C'].get_vector()
>>> ca = residue['CA'].get_vector()
# center at origin
>>> n = n - ca
>>> c = c - ca
# find rotation matrix that rotates n
# -120 degrees along the ca-c vector
>>> rot = rotaxis(-pi * 120.0/180.0, c)
# apply rotation to ca-n vector
>>> cb_at_origin = n.left_multiply(rot)
# put on top of ca atom
>>> cb = cb_at_origin+ca
这个例子展示了在原子数据上能进行一些相当不平凡的向量运算,这些运算会很有用。除了所有常用向量运算(叉积(用 ** ),点积(用 * ),角度, 取范数等)和上述提到的 rotaxis 函数,Vector 模块还有方法能旋转( rotmat )或反射( refmat )一个向量到另外一个向量上。
11.2.6 从结构中提取指定的 Atom/Residue/Chain/Model¶
举些例子如下:
>>> model = structure[0]
>>> chain = model['A']
>>> residue = chain[100]
>>> atom = residue['CA']
还可以用一个快捷方式:
>>> atom = structure[0]['A'][100]['CA']
11.3 紊乱¶
Bio.PDB能够处理紊乱原子和点突变(比如Gly和Ala残基在相同位置上)。
11.3.1 一般性方法¶
紊乱可以从两个角度来解决:原子和残基的角度。一般来说,我们尝试压缩所有由紊乱引起的复杂性。如果你仅仅想遍历所有Cα原子,那么你不必在意一些具有紊乱侧链的残基。另一方面,应该考虑在数据结构中完整地表示紊乱性。因此,紊乱原子或残基存储在特定的对象中,这些对象表现得就像毫无紊乱。这可以通过表示紊乱原子或残基的子集来完成。至于挑选哪个子集(例如使用Ser残基的哪两个紊乱OG侧链原子位置),由用户来决定。
11.3.2 紊乱原子¶
紊乱原子可以用普通的 Atom 对象来表示,但是所有表示相同物理原子的 Atom 对象都存储在一个 DisorderedAtom 对象中(见图. 11.1 )。 DisorderedAtom 对象中每个 Atom 对象都能用它的altloc标识符来唯一地索引。 DisorderedAtom 对象将所有未捕获方法的调用发送给选定的Atom对象,缺省对象是代表最高使用率的原子的那个。当然用户可以使用其altloc标识符来更改选定的 Atom 对象。以这种方式,原子紊乱就正确地表示出来而没有很多额外的复杂性。换言之,如果你对原子紊乱不感兴趣,你也不会被它困扰。
每个紊乱原子都有一个特征性的altloc标识符。你可以设定:一个 DisorderedAtom 对象表现得像与一个指定的altloc标识符相关的 Atom 对象:
>>> atom.disordered_select('A') # select altloc A atom
>>> print atom.get_altloc()
"A"
>>> atom.disordered_select('B') # select altloc B atom
>>> print atom.get_altloc()
"B"
11.3.3 紊乱残基¶
普通例子¶
最常见的例子是一个残基包含一个或多个紊乱原子。这显然可以通过用DisorderedAtom对象表示这些紊乱原子来解决,并将DisorderedAtom对象存储在一个Residue对象中,就像正常的Atom对象那样。通过将所有未捕获方法调用发送给其中一个Atom对象(被选定的Atom对象),DisorderedAtom对象表现完全像一个正常的原子对象(事实上这个原子有最高的使用率)。
点突变¶
一个特殊的例子就是当紊乱是由点突变导致的时候,也就是说,在晶体结构中出现一条多肽的两或多个点突变。关于这一点,可以在PDB结构1EN2中找到一个例子。
既然这些残基属于不同的残基类型(举例说Ser 60 和Cys 60),那么它们不应该像通常情况一样存储在一个单一 Residue 对象中。这种情况下每个残基用一个 Residue 对象来表示,两种 Residue 对象都保存在一个单一 DisorderedResidue 对象中(见图. 11.1 )。
DisorderedResidue 对象将所有未捕获方法发送给选定的 Residue 对象(默认是所添加的最后一个 Residue 对象),因此表现得像一个正常的残基。在 DisorderedResidue 中每个 Residue 对象可通过残基名称来唯一标识。在上述例子中,残基Ser 60在 DisorderedResidue 对象中的id为“SER”,而残基Cys 60则是“CYS”。用户可以通过这个id选择在 DisorderedResidue 中的有效 Residue 对象。
例子:假设一个链在位置10有一个由Ser和Cys残基构成的点突变。确信这个链的残基10表现为Cys残基。
>>> residue = chain[10]
>>> residue.disordered_select('CYS')
另外,通过使用 (Disordered)Residue 对象的 get_unpacked_list 方法,你能获得所有 Atom 对象的列表(也就是说,所有 DisorderedAtom 对象解包到它们各自的 Atom 对象)。
11.4 异质残基¶
11.4.1 相关问题¶
关于异质残基的一个很普遍的问题是同一条链中的若干异质和非异质残基有同样的序列标识符(和插入码)。因此,要为每个异质残基生成唯一的id,水分子和其他异质残基应该以不同的方式来对待。
记住Residue残基有一个元组(hetfield, resseq, icode)作为id。hetfield值为空(“ ”)表示为氨基酸和核酸;为一个字符串,则表示水分子和其他异质残基。hetfield的内容将在下面解释。
11.4.2 水残基¶
水残基的hetfield字符串由字母“W”构成。所以水分子的一个典型的残基id为(“W”, 1, “ ”)。
11.4.3 其他异质残基¶
其他异质残基的hetfield字符以“H_”起始,后接残基名称。一个葡萄糖分子,比如残基名称为“GLC”,则hetfield字符为“H_GLC”;它的残基id可以是(“H_GLC”, 1, “ ”)。
11.5 浏览Structure对象¶
解析PDB文件,提取一些Model、Chain、Residue和Atom对象¶
>>> from Bio.PDB.PDBParser import PDBParser
>>> parser = PDBParser()
>>> structure = parser.get_structure("test", "1fat.pdb")
>>> model = structure[0]
>>> chain = model["A"]
>>> residue = chain[1]
>>> atom = residue["CA"]
迭代遍历一个结构中的所有原子¶
>>> p = PDBParser()
>>> structure = p.get_structure('X', 'pdb1fat.ent')
>>> for model in structure:
... for chain in model:
... for residue in chain:
... for atom in residue:
... print atom
...
有个快捷方式可以遍历一个结构中所有原子:
>>> atoms = structure.get_atoms()
>>> for atom in atoms:
... print atom
...
类似地,遍历一条链中的所有原子,可以这么做:
>>> atoms = chain.get_atoms()
>>> for atom in atoms:
... print atom
...
遍历模型中的所有残基¶
或者,如果你想遍历在一条模型中的所有残基:
>>> residues = model.get_residues()
>>> for residue in residues:
... print residue
...
你也可以用 Selection.unfold_entities 函数来获取一个结构的所有残基:
>>> res_list = Selection.unfold_entities(structure, 'R')
或者获得链上的所有原子:
>>> atom_list = Selection.unfold_entities(chain, 'A')
明显的是, A=atom, R=residue, C=chain, M=model, S=structure 。你可以用这种标记返回层次中的上层,如从一个 Atoms 列表得到(唯一的) Residue 或 Chain 父类的列表:
>>> residue_list = Selection.unfold_entities(atom_list, 'R')
>>> chain_list = Selection.unfold_entities(atom_list, 'C')
更多信息详见API文档。
从链中提取异质残基(如resseq 10的葡萄糖(GLC)部分)¶
>>> residue_id = ("H_GLC", 10, " ")
>>> residue = chain[residue_id]
打印链中所有异质残基¶
>>> for residue in chain.get_list():
... residue_id = residue.get_id()
... hetfield = residue_id[0]
... if hetfield[0]=="H":
... print residue_id
...
输出一个结构分子中所有B因子大于50的CA原子的坐标¶
>>> for model in structure.get_list():
... for chain in model.get_list():
... for residue in chain.get_list():
... if residue.has_id("CA"):
... ca = residue["CA"]
... if ca.get_bfactor() > 50.0:
... print ca.get_coord()
...
输出所有含紊乱原子的残基¶
>>> for model in structure.get_list():
... for chain in model.get_list():
... for residue in chain.get_list():
... if residue.is_disordered():
... resseq = residue.get_id()[1]
... resname = residue.get_resname()
... model_id = model.get_id()
... chain_id = chain.get_id()
... print model_id, chain_id, resname, resseq
...
遍历所有紊乱原子,并选取所有具有altloc A的原子(如果有的话)¶
这将会保证,SMCRA数据结构会表现得如同只存在altloc A原子一样。
>>> for model in structure.get_list():
... for chain in model.get_list():
... for residue in chain.get_list():
... if residue.is_disordered():
... for atom in residue.get_list():
... if atom.is_disordered():
... if atom.disordered_has_id("A"):
... atom.disordered_select("A")
...
从 Structure 对象中提取多肽¶
为了从一个结构中提取多肽,需要用 PolypeptideBuilder 从 Structure 构建一个 Polypeptide 对象的列表,如下所示:
>>> model_nr = 1
>>> polypeptide_list = build_peptides(structure, model_nr)
>>> for polypeptide in polypeptide_list:
... print polypeptide
...
Polypeptide对象正是Residue对象的一个UserList,总是从单结构域(在此例中为模型1)中创建而来。你可以用所得 Polypeptide 对象来获取序列作为 Seq 对象,或获得Cα原子的列表。多肽可以通过一个C-N 化学键或一个Cα-Cα化学键距离标准来建立。
例子:
# Using C-N
>>> ppb=PPBuilder()
>>> for pp in ppb.build_peptides(structure):
... print pp.get_sequence()
...
# Using CA-CA
>>> ppb=CaPPBuilder()
>>> for pp in ppb.build_peptides(structure):
... print pp.get_sequence()
...
需要注意的是,上例中通过 PolypeptideBuilder 只考虑了结构的模型 0。尽管如此,还是可以用 PolypeptideBuilder 从 Model 和 Chain 对象创建 Polypeptide 对象。
获取结构的序列¶
要做的第一件事就是从结构中提取所有多肽(如上所述)。然后每条多肽的序列就容易从 Polypeptide 对象获得。该序列表示为一个Biopython Seq 对象,它的字母表由 ProteinAlphabet 对象来定义。
例子:
>>> seq = polypeptide.get_sequence()
>>> print seq
Seq('SNVVE...', <class Bio.Alphabet.ProteinAlphabet>)
11.6 分析结构¶
11.6.1 度量距离¶
重载原子的减法运算来返回两个原子之间的距离。
# Get some atoms
>>> ca1 = residue1['CA']
>>> ca2 = residue2['CA']
# Simply subtract the atoms to get their distance
>>> distance = ca1-ca2
11.6.2 度量角度¶
用原子坐标的向量表示,和 Vector 模块中的 calc_angle 函数可以计算角度。
>>> vector1 = atom1.get_vector()
>>> vector2 = atom2.get_vector()
>>> vector3 = atom3.get_vector()
>>> angle = calc_angle(vector1, vector2, vector3)
11.6.3 度量扭转角¶
用原子坐标的向量表示,然后用 Vector 模块中的 calc_dihedral 函数可以计算角度。
>>> vector1 = atom1.get_vector()
>>> vector2 = atom2.get_vector()
>>> vector3 = atom3.get_vector()
>>> vector4 = atom4.get_vector()
>>> angle = calc_dihedral(vector1, vector2, vector3, vector4)
11.6.4 确定原子-原子触点¶
用 NeighborSearch 来进行邻接查询。用C语言写的(使得运行很快)KD树模块(见 Bio.KDTree )可以用来完成邻接查询。它也包含了一个快速方法来找出相距一定距离的所有点对。
11.6.5 叠加两个结构¶
可以用 Superimposer 对象将两个坐标集叠加。这个对象计算出旋转和平移矩阵,该矩阵旋转两个列表上相重叠的原子使其满足RMSD最小。当然这两个列表含有相同数目的原子。 Superimposer 对象也可以将旋转/平移应用在一列原子上。旋转和平移作为一个元组储存在 Superimposer 对象的 rotran 属性中(注意,旋转是右乘),RMSD储存在属性 rmsd 中。
Superimposer 使用的算法来自[ 17 ,
Golub & Van Loan]并使用了奇异值分解(这是通用 Bio.SVDSuperimposer 模块中实现了的)。
例子:
>>> sup = Superimposer()
# Specify the atom lists
# 'fixed' and 'moving' are lists of Atom objects
# The moving atoms will be put on the fixed atoms
>>> sup.set_atoms(fixed, moving)
# Print rotation/translation/rmsd
>>> print sup.rotran
>>> print sup.rms
# Apply rotation/translation to the moving atoms
>>> sup.apply(moving)
为了基于有效位点来叠加两个结构,用有效位点的原子来计算旋转/平移矩阵(如上所述),并应用到整个分子。
11.6.6 双向映射两个相关结构的残基¶
首先,创建一个FASTA格式的比对文件,然后使用``StructureAlignment`` 类。这个类也可以用来比对两个以上的结构。
11.6.7 计算半球暴露(HSE)¶
半球暴露(Half Sphere Exposure,HSE)是对溶剂暴露 [ 20 ]的一种新的二维度量。根本上,它计数了围绕一个残基,在其侧链方向上及反方向(在13 Å范围内)的Cα原子。尽管简单,它表现得比溶剂暴露的其它度量都要好。
HSE有两种风味:HSEα和HSEβ。前者仅用到Cα原子的位置,而后者用到Cα和Cβ原子的位置。HSE度量是由 HSExposure 类来计算的,这个类也能计算触点数目。后一个类有方法能返回一个字典,该字典将一个``Residue`` 对象映射到相应的HSEα,HSEβ和触点数目值。
例子:
>>> model = structure[0]
>>> hse = HSExposure()
# Calculate HSEalpha
>>> exp_ca = hse.calc_hs_exposure(model, option='CA3')
# Calculate HSEbeta
>>> exp_cb=hse.calc_hs_exposure(model, option='CB')
# Calculate classical coordination number
>>> exp_fs = hse.calc_fs_exposure(model)
# Print HSEalpha for a residue
>>> print exp_ca[some_residue]
11.6.8 确定二级结构¶
为了这个功能,你需要安装DSSP(并获得一个对学术性使用免费的证书,参见 http://www.cmbi.kun.nl/gv/dssp/ )。然后用 DSSP 类,可以映射 Residue 对象到其二级结构上(和溶剂可及表面区域)。DSSP代码如下表所列表 11.1 。注意DSSP(程序及其相应的类)不能处理多个模型!
| Code | Secondary structure |
| H | α-helix |
| B | Isolated β-bridge residue |
| E | Strand |
| G | 3-10 helix |
| I | Π-helix |
| T | Turn |
| S | Bend |
| Other |
Table 11.1: Bio.PDB中的DSSP代码。
DSSP 类也可以用来计算残基的溶剂可及表面。还请参考 11.6.9 。
11.6.9 计算残基深度¶
残基深度是残基原子到溶剂可及表面的平均距离。它是溶剂可及性的一种相当新颖和非常强大的参数化。为了这个功能,你需要安装Michel Sanner的 MSMS程序( http://www.scripps.edu/pub/olson-web/people/sanner/html/msms_home.html )。然后使用 ResidueDepth 类。这个类像字典一样将 Residue 对象映射到相应的(残基深度,Cα深度)元组。Cα深度是残基的Cα原子到溶剂可及表面的距离。
例子:
>>> model = structure[0]
>>> rd = ResidueDepth(model, pdb_file)
>>> residue_depth, ca_depth=rd[some_residue]
你也可以以带有表面点的数值Python数组的形式获得分子表面本身(通过 get_surface 函数)。
11.7 PDB文件中的常见问题¶
众所周知,很多PDB文件包含语义错误(不是结构本身的错误,而是在PDB文件中的表示)。Bio.PDB可以有两种方式来处理这个问题。PDBParser对象能表现出两种方式:严格方式和宽容方式(默认方式):
例子:
# Permissive parser
>>> parser = PDBParser(PERMISSIVE=1)
>>> parser = PDBParser() # The same (default)
# Strict parser
>>> strict_parser = PDBParser(PERMISSIVE=0)
在宽容状态(默认),明显包含错误的PDB文件会被“纠正”(比如说一些残基或原子丢失)。这些错误包括:
- 多个残基使用同一个标识符
- 多个原子使用统一个标识符(考虑altloc识别符)
这些错误暗示了PDB文件中确实存在错误(详情见 [18, Hamelryck and Manderick, 2003] )。在严格模式,带错的PDB文件会引发异常,这有助于发现PDB文件中的错误。
但是有些错误能自动修正。正常情况下,每个紊乱原子应该会有一个非空altloc标识符。可是很多结构没有遵循这个惯例,而在同一原子的两个紊乱位置存在一个空的和一个非空的标识符。这个错误会被以正确的方式自动解析。
有时候一个结构会有这样的情况:一部分残基属于A链,接下来一部分残基属于B链,然后又有一部分残基属于A链,也就是说,这种链是“断的”。这也能被自动正确解析。
11.7.1 例子¶
PDBParser/Structure类经过了将近800个结构(每个都属于不同的SCOP超家族)上的测试。测试总共耗时20分钟左右,或者说平均每个结构只需1.5秒。在一台1000 MHz的PC上只需10秒就可解析包含近64000个原子的大核糖体亚基(1FKK)的结构。
当不能建立明确的数据结构时会发生三类异常。在这三类异常中,可能的起因是PDB文件中一个本应修正的错误。这些情况下产生异常要比冒险地错误描述一个数据结构中的结构好得多。
11.7.1.1 重复残基¶
一个结构包含在一条链中具有相同的序列标识符(resseq 3)和icode的两个氨基酸残基。仔细观察可以发现这条链包含残基:Thr A3, …, Gly A202, Leu A3, Glu A204。很明显第二个Leu A3应该是Leu A203。类似的情况也存在于结构1FFK(比如它包含残基Gly B64, Met B65, Glu B65, Thr B67,也就是说Glu B65应该是Glu B66)上。
11.7.1.2 重复原子¶
结构1EJG含有在A链22位的一个Ser/Pro点突变。依次,Ser 22含一些紊乱原子。和期望的一样,所有属于 Ser 22的原子都有一个非空的altloc标识符(B或C)。所有Pro 22的原子都有altloc A,除了含空altloc的N原子。这会生成一个异常,因为一个点突变处属于两个残基的所有原子都应该有非空的altloc。结果这个原子很可能被Ser 和 Pro 22共用,而Ser22丢失了这个N原子。此外,这也点出了文件中的一个问题:这个N原子应该出现在Ser和Pro残基中,两种情形下都与合适的altloc标识符关联。
11.7.2 自动纠正¶
一些错误相当普遍且能够在没有太大误解风险的情况下容易地纠正过来。这些错误列在下面。
11.7.2.1 紊乱原子的空altloc¶
正常情况下,每个紊乱原子应该会有一个非空altloc标识符,可是很多结构没有遵循这个惯例,而是在同一原子的两个紊乱位置存在一个空的和一个非空的标识符。这个错误会被以正确的方式自动解析。
11.7.2.2 断链¶
有时候一个结构会有这样的情况:一部分残基属于A链,接下来一部分残基属于B链,然后又有一部分残基属于A链,也就是说,链是“断的”,这也能被正确的解析。
11.7.3 致命错误¶
有时候一个PDB文件不能被明确解释。这会产生异常并等待用户去修正这个PDB文件,而不是猜测和冒出错的风险。这些异常列在下面。
11.7.3.1 重复残基¶
在一条链上的所有残基都应该有一个唯一的id。该id基于下述生成:
- 序列标识符(resseq)
- 插入码(icode)
- hetfield字符(“W”代表水,“H_”后面的残基名称代表其他异质残基)
- 发生点突变的残基的名称(在DisorderedResidue对象中存储Residue对象)
如果这样还不能生成一个唯一的id,那么肯定是一些地方出了错,这时会生成一个异常。
11.7.3.2 重复原子¶
一个残基上所有原子应该有一个唯一的id,这个id基于下述产生:
- 原子名称(不带空格,否则会报错)
- altloc标识符
如果这样还不能生成一个唯一的id,那么肯定是一些地方出了错,这时会生成一个异常。
11.8 访问Protein Data Bank¶
11.8.1 从Protein Data Bank下载结构¶
结构可以从PDB(Protein Data Bank)通过 PDBList 对象的 retrieve_pdb_file 方法下载。这种方法的要点是结构的PDB标识符。
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('1FAT')
PDBList 类也能用作命令行工具:
python PDBList.py 1fat
下载的文件将以 pdb1fat.ent 为名保存在当前工作目录。注意 retrieve_pdb_file 方法还有个可选参数 pdir 用来指定一个特定的路径来保存所下载的PDB文件。
retrieve_pdb_file 方法还有其他选项可以指定下载所用的压缩格式(默认的 .Z 格式和 gunzip 格式)。另外,在创建 PDBList 对象时还可以指定PDB ftp站点。默认使用Worldwide Protein Data Bank( ftp://ftp.wwpdb.org/pub/pdb/data/structures/divided/pdb/ )。详细内容参见API文档。再次感谢Kristian Rother对此模块的所做的贡献。
11.8.2 下载整个PDB¶
下面的命令将会保存所有PDB文件至 /data/pdb 目录:
python PDBList.py all /data/pdb
python PDBList.py all /data/pdb -d
在API中这个方法叫做 download_entire_pdb 。添加 -d 会在同一目录下保存所有文件。否则将分别保存至PDB风格的、与其PDB ID对应的子目录中。根据网速,完整的下载全部PDB文件大概需要2-4天。
11.8.3 保持本地PDB拷贝的更新¶
这也能通过 PDBList 对象来完成。可以简单的创建一个 PDBList 对象(指定本地PDB拷贝的目录),然后调用 update_pdb 方法:
>>> pl = PDBList(pdb='/data/pdb')
>>> pl.update_pdb()
当然还可以每周用 cronjob 实现本地拷贝自动更新。还可以指定PDB ftp站点(详见API文档)。
PDBList 有其他许多其它方法可供调用。 get_all_obsolete 方法可以获取所有已经废弃的PDB项的一个列表; changed_this_week 方法可以用于获得当前一周内新增加、修改或废弃的PDB项。更多 PDBList 的用法参见API文档。
11.9 常见问题¶
11.9.1 Bio.PDB测试得如何?¶
事实上,相当好。Bio.PDB已经在从PDB获得的近5500个结构上广泛的测试过,所有结构都能正确地解析。更多细节可以参考在Bioinformatics上发表的关于Bio.PDB的文章。作为一个可靠的工具,Bio.PDB已经并正用于许多研究项目中。我几乎每天都在用它,出于研究目的、提升其性能和增加新属性。
11.9.2 它有多快?¶
PDBParser 的性能经过将近800个结构测试(每个都属于不同的SCOP超家族),总共花费20分钟左右,也就是说平均每个结构只需1.5秒。在一台1000 MHz的PC上解析巨大的包含近64000个原子的核糖体亚单位(1FKK)只需10秒。总而言之,它比很多应用程序都快得多。
11.9.3 是否支持分子图形展示?¶
不直接支持,很大程度上是因为已有相当多基于Python或Python-aware的解决方案,也可能会用到Bio.PDB。顺便说一下,我的选择是Pymol(我在Pymol中使用Bio.PDB非常成功,将来Bio.PDB中会有特定的PyMol模块)。基于Python或Python-aware的分子图形解决方案包括:
- PyMol: http://pymol.sourceforge.net/
- Chimera: http://www.cgl.ucsf.edu/chimera/
- PMV: http://www.scripps.edu/~sanner/python/
- Coot: http://www.ysbl.york.ac.uk/~emsley/coot/
- CCP4mg: http://www.ysbl.york.ac.uk/~lizp/molgraphics.html
- mmLib: http://pymmlib.sourceforge.net/
- VMD: http://www.ks.uiuc.edu/Research/vmd/
- MMTK: http://starship.python.net/crew/hinsen/MMTK/
11.9.4 谁在用Bio.PDB?¶
Bio.PDB曾用于构建DISEMBL,一个能预测蛋白结构中的紊乱区域的web服务器( http://dis.embl.de/ );COLUMBA,一个提供注释过的蛋白结构的站点( http://www.columba-db.de/ )。Bio.PDB也用于进行PDB中蛋白质间有效位点的大规模相似性搜索 [19, Hamelryck, 2003] ,用于开发新的算法来鉴别线性二级结构元件 [26, Majumdar et al., 2005] 。
从对特征和信息的需求判断,许多大型制药公司也使用Bio.PDB。